样本表达量质量评估(剔除离群样本)

图片alt
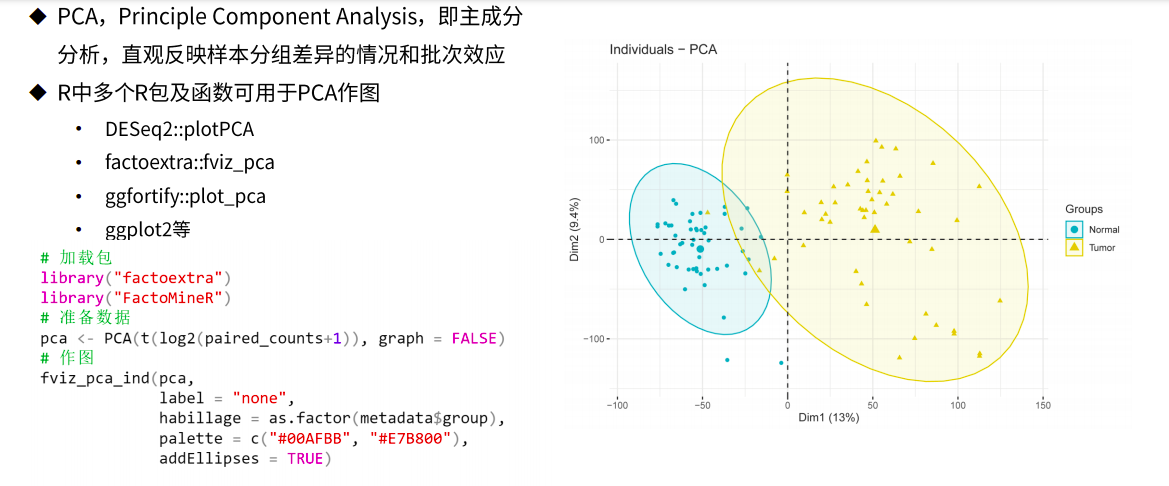
图片alt

图片alt
提取配对样本
metadata <- count_obj@metadata %>%
filter(tissue_type_id %in% c("01", "11")) %>%
add_count(patient_id, name = "n_patient")%>%
filter(n_patient == 2) %>%
arrange(patient_id,tissue_type_id) %>%
as.data.frame()
group <- metadata$group %>% as.factor()
paired_counts <- count_obj@count[,metadata$cases]
keep <- rowSums(paired_counts > 0) >= 38
paired_counts <- paired_counts[keep,]
配对样本质量控制
boxplot(log2(paired_counts+1), las = 2, outline = F, col = group)
limma::plotDensities(log2(paired_counts+1), legend = T,group=group,col = c("green","red"))

图片alt
vst转换配对样本质量控制
library(DESeq2)
group <- metadata$group %>% as.factor()
colData = data.frame(sample_id = colnames(paired_counts), group = group)
DDS <- DESeq2::DESeqDataSetFromMatrix(paired_counts,
colData = metadata,
design = ~ group)
vst <- DESeq2::vst(DDS)
counts_vst <- assay(vst)
boxplot(counts_vst, las = 2, outline = F, col = group)
limma::plotDensities(counts_vst , legend = F,group=metadata$group)
过滤基因
keep <- rowSums(paired_counts > 0) >= 38
expr <- lnRAN_mRNA(fpkm_obj)@lnRNA[lnRNA_deg_sig$symbol,]
keep <- rowSums(expr > 0.5)>=2 # keep <- rowSums(cpm(y) > 0.5) >= 2
table(keep)
keep <- filterByExpr(exprSet,group = group)
DeSeq2差异表达分析
limma包差异表达分析
makeContrasts(contrasts=cts, levels=design)1 比较的 -1 被比较的

图片alt
edgeR差异基因分析
# edgeR差异基因分析
library(edgeR)
group <- c(rep(1,length(Norm_sample)),rep(2,length(Tumm_sample)))
y <- DGEList(counts=count_order,group=group)
# 数据过滤
keep <- filterByExpr(y)
y <- y[keep,,keep.lib.size=F]
# 计算标准化因子
y <- calcNormFactors(y)
# 计算离散度
y <- estimateDisp(y)
# 显著性检验
et <- exactTest(y)
# 获取靠前的基因
et <- topTags(et,n=100000)
# 转换为数据框
et <- as.data.frame(et)
# 将行名粘贴为第一列
et <- cbind(rownames(et),et)
str(et)
ggplot(et,aes(x=logFC,y=-log10(FDR)))+
geom_point()
参考
https://www.jianshu.com/p/e68c95a42cd6
https://www.jianshu.com/p/f009bea514af?utm_campaign=haruki