摘 要:连翘是传统的中药材,有着极高的经济价值,被商洛市确定的“十大商药”主要产品之一。为从分子角度去理解连翘苷和连翘酯苷A在叶子和果实中的差异,本研究通过采用RNA-Seq测序技术对连翘的转录组进行测序,并拼接转录本,得到142 098条Unigene。同时对其基因进行了功能注释,发现大量与萜类骨架生物合成途径、苯丙烷生物合成途径、二萜生物合成,类胡萝卜素生物合成等多个次生代谢相关的代谢通路。此外本研究也比较了连翘叶和果实中的差异基因,并且对差异基因进行富集分析,发现连翘叶子和果实中次生代谢物质含量不同可能与苯丙烷代谢途径、类胡萝卜素生物合成有关。
关键词:连翘;转录组;功能注释;差异表达
Abstract:Forsythia suspensa is a traditional Chinese medicinal material with extremely high economic value. It is one of the main products of the "Ten Major Commercial Medicines" identified by Shangluo City. In order to understand the differences between the main components of F. suspensa in leaves and fruits from a molecular perspective, we sequenced the transcriptome of F. suspensa by using RNA-Seq sequencing technology and spliced the transcripts to obtain 142 098 unigenes. At the same time, its genes were functionally annotated, and a large number of genes related to secondary metabolism were found. In addition, we also compared the differential genes in the leaves and fruits of F. suspensa, and analyzed the enrichment of the differential genes, and found that the different content of secondary metabolites in the leaves and fruits of F. suspensa may be related to the phenylpropane metabolic pathway and carotenoid biosynthesis.
Key words: Forsythia suspensa; transcriptome; functional annotation; differential expression
引言
连翘(Forsythia suspensa (Thunb.) Vahl)被商洛市定为“十大商药”之一[1],蕴藏量大,分布区域广,在商洛拥有悠久的种植历史及广泛的种植面积,其作用主要是清热解毒,散结消痈,疏散风热[2],现代药理学研究表连翘具有显著的抗炎、抗病毒等作用[3]。其中连翘苷和连翘酯苷A作为连翘主要成分的代表,可在干燥果实和叶中的提取,是作为连翘有效成分的判定指标[2],如在中国药典中就有规定,在对连翘药材质量进行鉴定时连翘苷含量不得低于0.15%,青翘含连翘酯苷A不得少于3.5%;老翘含连翘酯苷A不得少于0.25%。在叶片与果实的有效成分的含量鉴定中,目前已有论文研究连翘果实和叶片提取物对大肠杆菌、金黄色葡萄球菌、枯草芽孢杆菌抑菌效果中,叶片的提取物均大于果实的提取物[4]。连翘的不同部位,连翘叶中含有连翘苷、连翘酯苷A含量远高于常作为药材原料入药的连翘果中的含量,其具有较高的潜在药用价值[5][6]。连翘不同部位炮制,连翘茎叶蒸晒品中含有较高含量的连翘酯苷,以连翘叶作为茶叶开发具有广阔前景[7]。但是目前并没有从分子角度去阐明连翘果实及叶片中这两种次生代谢物质含量出现差别的机制。因此从分子调控去了解连翘果实和叶子次生代谢机制是有必要的。
目前,对次生代谢调控网络及其基因相关功能的认知是药用植物次生代谢工程中需要解决的最大问题。次生代谢产物是药用植物的主要成分,但是次生物质代谢途径相对比较其复杂,其过程受到许多基因的调控。由于缺少全基因组序列数据,深入的分子调控及与次生代谢相关基因研究几乎没有展开。本文通过采用转录组RNA-Seq测序技术,完成了4个连翘样品(2个叶片样品,2个果实样品)的转录组测序工作,并完成了Unigene的拼接和功能注释。使用注释的基因对连翘果实和叶片的表达量进行差异基因表达分析,并且对差异表达基因进行KEGG和GO富集分析,进而从分子角度去理解连翘不同部位(叶片和果实)中次生代谢的调控机制。
转录组(Transcriptome)是特定类型细胞中全体转录本的一个集合。狭义上,转录组是指所有与蛋白质翻译相关的mRNA;广义上的转录组指DNA转录的所有mRNA,不仅包含编码蛋白mRNA也包含非编码蛋白的miRNA、lncRNA和其他ncRNA。目前转录组研究技术主要包括两类:一类是基于杂交技术的Microarray;另一类是基于测序的RNA测序技术(RNA-seq )、表达序列标签技术(EST)等。基于杂交的Microarray技术和基于测序的RNA-seq技术都可以对样本中的所有转录本进行定性和定量分析,所谓定性分析是指鉴定表达的基因,定量分析是指获得基因的表达量,但是Microarray技术需要事先知道待检测转录本的序列,因此不能发现新的基因;而RNA-seq技术不仅可以发现新的基因还能够发现转录组中所有的可变剪切,但是检测的效率和灵敏度高度依赖测序的深度,因此对测序数据进行质量评估是转录组数据分析的重要环节。
使用RNA-Seq对转录组进行测序已经广泛用于药用植物基因组的研究当中。RNA-Seq技术能够对不同组织类型、不同处理条件、不同生理状态、不同时间点、野生型和突变型等,不同条件下调控方式与基因表达的差异进行研究,与此同时RNA-Seq技术可以快速获取非模式药用植物的转录本信息,并且对基因的编码序列进行预测。因此可利用RNA-Seq技术研究转录本的结构和变异。
1 材料与方法
1.1 植物材料
4个连翘样品(2个叶片样品,2个果实样品)采自陕西商洛市。在8月份,分别收获叶片和果实,每个样品取两个作为重复。
1.2 RNA分离和测序
使用上海生工生物工程股份有限公司的 Trizol 试剂盒(Total RNA Extractor SK1312),提取流程按照说明书严格操作。分别提取连翘叶子和果实的总RNA,获得cDNA文库之后,使用Illumina Hi-Seq 2500平台进行双末端测序。
1.3 质量控制及转录组组装
测序得到Raw Data除去其中rRAN、接头和低质量的reads,使得Q30(碱基的测序错误率小于 0.1%)达到 80%以上。从而得到干净数据(clean reads),当检测reads的质量符合要求之后,可以使用Trinity软件[8]进行转录组进行据组装。为了确保转录本组装的完整性,本研究我们采用同物种合并组装。Trinity软件的基本操作流程是,根据测序得到的Reads生成K-mer库;以次数最多的K-mer为种子生成得到contig;对Contig进行聚类,构建聚类得到集群的de Bruijn图;以测序的reads解开De Bruijn图进行简化。获得转录本序列。N50是转录组组装完整度的指标之一,是指先将所有片段按照从长到短的顺序排序,再从第一条片段开始累加,当相加达到总长度50%时对应的那条片段的长度。一般认为N50长度≥800bp时组装的序列完整性较好。
1.4 Unigene功能注释
使用BLASTx[9]软件将Unigene序列分别与数据库NR[10]、Swiss-Prot[11]、GO[12]、eggNOG4[13]、 KEGG[14]等进行比对,使用KOBAS2.0[15]分析Unigene在KEGG数据库中的注释结果;当Unigene的氨基酸序列被预测之后,使用软件HMMER[16]与Pfam数据库进行比对,进而得到Unigene的注释信息。
1.5 差异表达基因分析及富集分析
基因的在不同的时间和空间表达量不同,当在不同处理之下的差异具有统计学意义的基因,被称为差异表达基因。选择连翘不同部位及果实和叶子中基因的表达量,使用DESeq[17],进行差异表达分析。以FDR(False Discovery Rate)小于0.01且差异倍数FC(Fold Change)大于等于2为筛选条件,得到差异基因。接着对差异的基因进行GO富集分析和KEGG富集分析。即寻找在某一个KEGG通路或者GO术语上是否出现over-presentation。本文采用富集因子(Enrichment Factor)分析差异基因在KEGG通路中的富集程度,在计算差异基因的富集显著性时,采用Fisher统计检验。
2 结果与分析
2.1 转录组测序数据组装
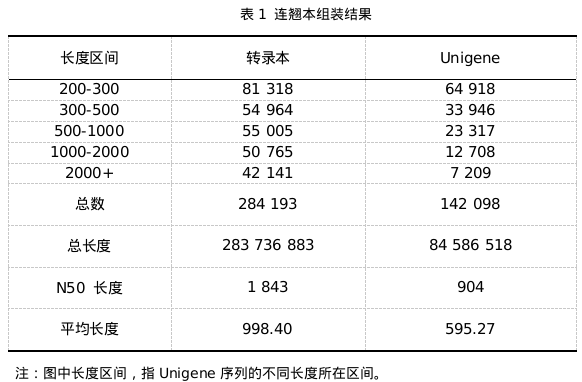
采用转录组RNA-Seq测序技术对4个连翘样品(2个叶片样品,2个果实样品)进行测序,在去除接头及低质量值数据后,得到4个样品reads分别为34 139 187、27 620 447、31 280 295、28 424 671,共36.09Gb,GC含量分别为44.27%、44.46%、44.25%、44.48%,测序得到碱基错误率,即Q30分别为91.27%、91.44%、91.84%、91.53%,说明测序质量较好,可以作为后续组装和分析的输入数据(表1)。
使用Trinity对转录本进行组装,本研究得到142,098条Unigene,平均长度为595.27 nt,总长度为84 586 518 nt,其中长度在200~300 nt的Unigene 最多,达64 918(45.69%)个(表1),其次是300~500nt的Unigene33 946个(23.89%),500~1000nt的Unigene 有23 317个(16.41%),1000~ 2000nt的Unigene有112 708个(8.94%),长度大于2000nt的Unigene为7 209(5.07%)条。转录本的unigene N50长度为904nt,表明组装片段的完整性比较高。
图片alt
2.2 Unigene功能注释
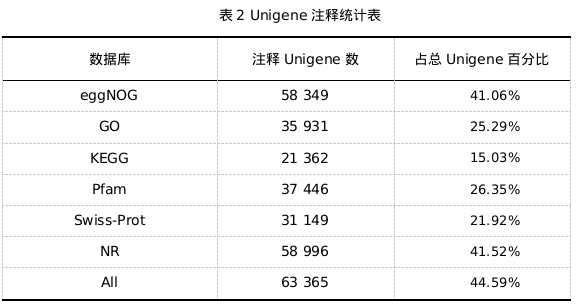
对连翘的基因功能进行注释和分类使用Blast的方法,其结果见表2。Unigenes中有43.98% (62 496个)的基因得到了注释。其中在NCBI中的非冗余蛋白质数据库NR数据库中注释的基因最多,有58 996条;在由EBI(欧洲生物信息学研究所)负责维护的数据库Swiss-Prot注释到31 149条,其可信度相对较高;在Pfam(Protein family)通过氨基酸序列预测蛋白质的功能的数据库中,有37 446个基因注释到该数据库。有79602条unigenes没有得到注释,注释结果中存在超过1/2的unigenes没有得到注释,这与连翘缺少参考基因组有关。总的来说,62 496个Unigenes可以通过与七个数据库注释中的至少六个数据库来注释(图1)。
图片alt
注:数据库,表示对Unigene进行预测的数据库;注释Unigene数,表示在对应数据库中注释得到的Unigene个数;占总Unigene百分比,每个数据库注释的Unigene数占所有组装得到Unigene数的百分比。

图片alt
注:图中标有数字的交集区域表示,交集的数据库共同注释的Unigene个数;没有标数字的区域表示,该数据库注释的Unigene都可以在其它数据库得到注释。
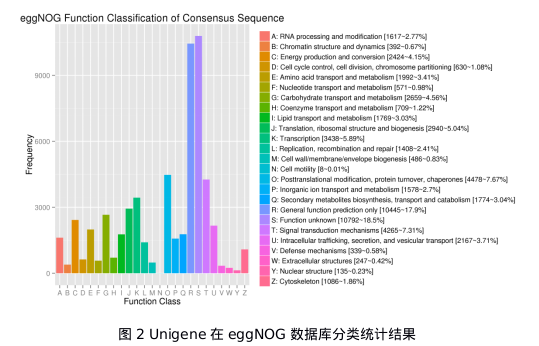
eggNOG是一个对直系同源蛋白的功能和分类的数据库,结合了COG(Clusters of Orthologous Groups)识别直系同源基因的数据库,KOG(euKaryotic Orthologous Groups)根据真核生物基因直系同源关系和进化关系而构建的数据库。在eggNOG数据中注释结果显示(图2)注释的Unigene有58 349个,共分为25个类别。其中大多数基因注释到一般功能预测10 445(17.90%),其次是翻译后修饰、蛋白质周转、分子伴侣4 478个(7.67%)、信号转导机制4 265个(7.31%)、转录3 438个(5.89%)、翻译、核糖体结构和生物发生2 940个(5.04%)、碳水化合物运输和代谢2 659个(4.56%)、能源生产和转化2 424个(4.15%)、细胞内运输、分泌和囊泡运输2 167个(3.71%)、氨基酸转运和代谢1 992个(3.41)。有10 792个Unigene 功能未知,这些Unigene很可能是连翘的特异基因。此外参与次级代谢物的生物合成、运输和分解代谢的基因有1 774个(3.04 %),对这些基因进一步研究可以帮助了解连翘有效成分合成的分子机制。
图片alt
注:横标表示eggNOG数据库注释分类名称;纵坐标表示某个分类上注释到的Unigene数目
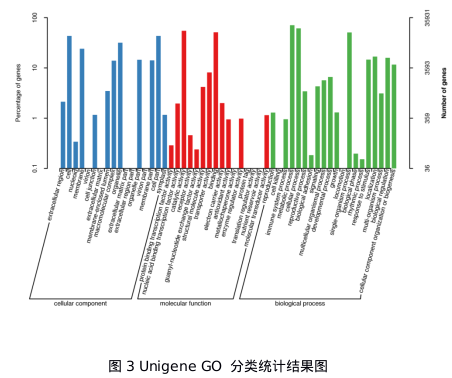
GO数据库对基因的序列进行了结构化和标准化的定义,对基因和基因产物的描述建立了一套标准的词库。该数据库是一个多层级的数据库,越低的节点代表的功能越具体。在GO数据库注释结果显示(图3)注释的Unigene有35931个,在细胞组分、生物学过程、分子功能注释的Unigene分别为69 660、45 462、96 645。进一步对Unigene进行二级功能分类,其中,代谢进程(25 376)、细胞进程(22 026)、 催化活性(19 683)、结合活性(18 409)、单一生物过程(18 194)、细胞(15 602)、细胞部分(15 602)和细胞器(11 438)功能组中涉及的Unigene较多,而胞外区部分(9)、细胞外基质(8)、翻译调节器活性(7)、细胞杀伤(6)、蛋白质标签(4)、细胞外基质(1)功能组中涉及的Unigene 较少。
图片alt
注:图中的蓝色、红色、绿色柱状图表示Unigene分别注释到细胞组分、分子功能、生物学过程对应的二级分类功能的数目。
KEGG数据库是系统分析代谢网络的数据库。在KEGG数据库中注释结果显示注释Unigene有21 362个,分类于130条代谢通路。其中最多的是核糖体途径1 280个(5.99%),其次是碳代谢1 211个(5.67%),氨基酸的生物合成1 021个(4.78%),内质网中的蛋白质加工741个(3.34%),氧化磷酸化661个(3.09%)(图4),而花青素生物合成、异黄酮生物合成、万古霉素耐药、碳青霉烯生物合成、黄酮和黄酮醇的生物合成、聚酮糖单元生物合成、甜菜碱生物合成途径相关基因较少,分别为3、3、3、2、2、1个。注释到次生代谢途径的Unigene在苯丙烷生物合成途径包括274个、萜类骨架生物合成途径包括171个,泛醌和其他萜类-醌生物合成包括89个,二萜生物合成包括38个,类胡萝卜素生物合成包括53个,说明连翘次生代谢途径的多样性和复杂性,这将为后续阐明其主要成分合成提供理论依据。
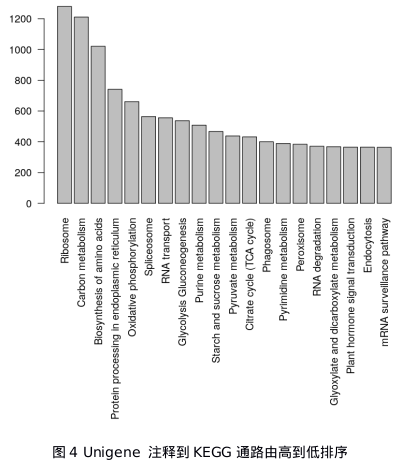
图片alt
注:图中横坐标表示KEGG通路描述;纵坐标表示注释到KEGG数据库的Unigene个数
NR数据库可以对同源序列的功能注释,同时也可以预测相近的物种。由(图5)可以发现芝麻与连翘同源性最高的是,其次是苜蓿。
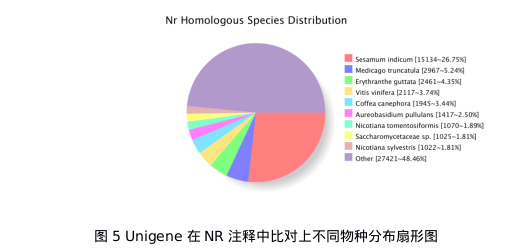
图片alt
注:图中不同的颜色对应不同的物种名称;每一个不同颜色所占扇形区域表示,注释到某物种的基因占总检测到的基因的比例
差异表达基因分析
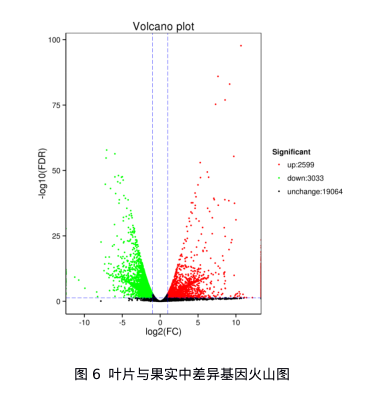
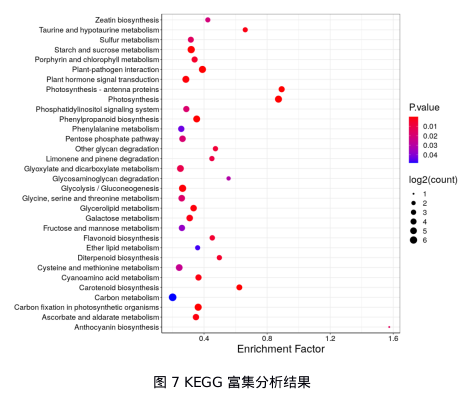
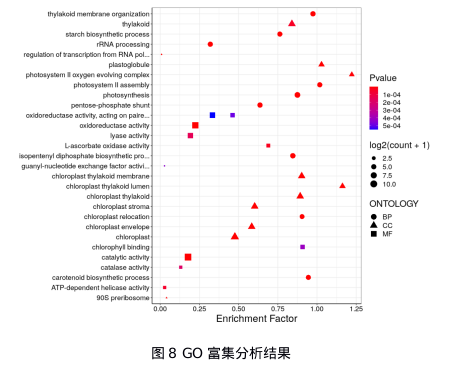
以FDR(False Discovery Rate)小于0.01且差异倍数FC(Fold Change)大于等于2作为筛选标准,筛选差异基因,结果(图6)在连翘的叶子和果实之间获得差异基因5632个,占总数的3.96%。与果实相比,在叶子中有2599个基因表达上调,3033个基因表达下调,其中有1578个基因注释到KEGG数据库,并且归属122条KEGG代谢途径。以所有的注释到KEGG的Unigene数目(35 319个)为背景,对差异基因进行富集分析。KEGG富集分析结果(图7)显示差异基因主要富集在光合作用、光合作用-天线蛋白、类胡萝卜素生物合成、牛磺酸和亚牛磺酸代谢、二萜生物合成、苯丙烷生物合成、淀粉与蔗糖代谢、玉米素生物合成、甘油脂代谢、糖酵解/糖异生、光合生物中的碳固定、卟啉与叶绿素代谢等生物学途径。GO富集结果(图7)显示,在生物学过程(biological process)富集的GO术语有类囊体膜组织、异戊烯二磷酸生物合成过程,光系统II组装、RNA聚合酶II启动子转录的调控、类胡萝卜素生物合成过程、淀粉生物合成过程、磷酸戊糖分流、光合作用、叶绿体搬迁、rRNA加工等。在分子功能(molecular function)富集的GO术语有氧化还原酶活性、催化活性、L-抗坏血酸氧化酶活性、ATP依赖性解旋酶活性、裂解酶活性、过氧化氢酶活性、叶绿素结合、鸟苷酸交换因子活性等。在细胞组分(cellular component)富集的GO术语有叶绿体类囊体膜、叶绿体基质、叶绿体被膜、叶绿体类囊体腔、叶绿体、质体小球、叶绿体类囊体、90S前核糖体、光系统II氧进化复合物、类囊体等。
图片alt
注:横坐标表示差异倍数的对数值;纵坐标表示错误发现率的负对数值,该值越大,表明假阳性的概率越低,该基因有较大的可能是生物学上的差异基因;图中有显著差异的基因用绿色和红色点表示,其中上调基因用红色表示,下调基因用绿色表示。
图片alt
注:图中纵坐标标表示KEGG通路名称;横坐标表示富集因子,其值越大,表示该基因有较大的可能在对应的通路中发挥作用;图例中的count即圆圈的大小表示该通路中差异基因的个数,越大圆圈表示差异基因在该通路的个数越多;图例中P-value为经过fisher检验后假阳性的概率,颜色越偏红色表示假阳性概率越低。
图片alt
注:图中纵坐标标表示GO术语;横坐标表示富集因子,其值越大,表示该基因越有可能在该GO术语中;图例中的count即圆圈的大小表示该GO术语中差异基因的个数,越大的圆圈表示差异基因在该GO术语的个数越多;图例中P-value为经过fisher检验后假阳性的概率,颜色越偏红色表示假阳性概率越低;图例中ONTOLOGY分别用圆形、三角形、正方形描述GO术语在不同分类BP生物学过程、CC细胞组分、MF分子功能中的富集情况。
3 讨论
连翘为常用的且市场需求量大的中药材,其规范化种植、生产的研究目前主要集中于水肥、种植及采收等方面,而由于缺少全基因组序列数据,深入的分子调控及与次生代谢相关基因研究几乎没有展开。RNA-seq作为非模式生物基因功能的研究、新基因的发现的快捷途径。转录组测序技术已经应用到许多常见的药用植物当中,并且获得大量功能基因,并且用这些功能基因解决实际的生物学问题。如贺润丽[18]等对款冬不同部位进行转录组测序,获得91 118个Unigene。本研究采用连翘苷和连翘酯苷A含量差异较大的连翘叶和果实进行比较转录组分析,得到142 098条unigene。通过对Unigene进行注释,发现许多与次生代谢相关的基因被包含在其中,例如在萜类骨架生物合成途径中包含的基因有171个,在类胡萝卜素生物合成途径的包含的基因有53个,在苯丙烷生物合成途径中包含的基因有274个,在泛醌和其他萜类-醌生物合成途径中包含的基因有89个,在二萜生物合成途径的包含的基因有38个。深入研究这些与次生代谢相关的基因有助于明确连有翘主要成分的分子机制,为从事连翘相关的科研工作者提供方便。但是这个基因功能的注释来自计算机的统计假设推断,需要进一步进行基因功能的研究。
通过比较连翘叶子和果实中的差异基因,并且对其中的差异基因进行富集分析。发现这些差异基因主要富集在光合作用、类胡萝卜素生物合成、二萜生物合成、苯丙烷生物合成、淀粉与蔗糖代谢等生物学途径。研究表明其中苯丙烷代谢途径是重要的植物次生代谢途径,苯丙烷代谢不仅能够产生松柏醇作为合成连翘苷的前体物,同时也能合成咖啡酰辅酶A作为连翘酯苷A的前体物质[19]。类胡萝卜素是光合色素之一,是一种四萜类的化合物,作用于光能的吸收与能量的传递。在逆境中,类胡萝卜素通过抑制叶绿体产生三线态叶绿素,进而具有光化学保护功能[20]。这说明类胡萝卜素生物合成与苯丙烷生物合成在连翘次生代谢中的重要作用。
4 结论
本研究从头组装了非模式生物连翘的转录组序列,并且对其基因进行了功能注释,发现大量与次生代谢相关的基因。此外我们也比较了连翘苷和连翘酯苷A含量差异较大的连翘叶和果实中的差异基因,并且对差异基因进行富集分析,发现连翘叶子和果实中次生代谢物质含量不同可能与苯丙烷代谢途径、类胡萝卜素生物合成有关。
参考文献
[1] 王超锋,曹小军.论陕西商洛市中药材产业发展[J].商洛学院学报,2021,35(01):92-96.
[2] 国家药典委员会.中华人民共和国药典: 一部[M].北京:中国医药科技出版社, 2015:170–171.
[3] 马忠英, 张迪, 孙金, 等. 连翘酯苷A通过TLR4/NF-κB抑制PC12细胞低氧/再复氧诱导的炎症反应[J].安徽医科大学学报,2021,56(05):730-734.
[4] 王小敏, 陈乔, 郭丽丽, 等. 连翘果、连翘叶乙醇提取物的抑菌活性及成分分析[J]. 食品工业科技, 2019, 40(06): 89-94.
[5 ]王进明, 范圣此, 李安平,等. 连翘不同部位中连翘苷和连翘酯苷A的含量分析及其入药探讨[J]. 中国现代中药, 2013, 15(07): 556-559.
[6] 李发荣, 段飞, 杨建雄. 中药连翘及连翘叶中连翘苷含量的比较研究[J]. 西北植物学报, 2004(04): 725-727.
[7] 周改莲, 辛宁, 张守平, 等. 连翘不同部位及不同炮制方法的活性成分比较[J]. 中国中医药信息杂志, 2008(S1): 30-31.
[8] Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome [J]. Nat Biotechnol. 2011, 29(7): 644-652.
[9] Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs [J]. Nucleic Acids Res. 1997, 25(17): 3389-402.
[10] Deng YY, Li JQ, Wu SF, et al. Integrated nr Database in Protein Annotation System and Its Localization [J]. Computer Engineering Italic. 2006. 32(5):71-74
[11] Apweiler R, Bairoch A, Wu CH, et al. UniProt: the Universal Protein knowledgebase [J]. Nucleic Acids Research Italic. 2004. 32(Database issue):D115-119.
[12] Ashburner M, Ball C A, Blake J A, et al. Gene ontology: tool for the unification of biology [J]. Nature genetics Italic. 2000. 25(1): 25-29.
[13]Huerta-Cepas J, Szklarczyk D, Forslund K, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences [J]. Nucleic Acids Res. 2016, 44(D1): D286-293.
[14] Kanehisa M, Goto S, Kawashima S, et al. The KEGG resource for deciphering the genome [J]. Nucleic Acids Research Italic. 2004. 32(Database issue): D277-D280.
[15] Xie C, Mao X, Huang J, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases [J]. Nucleic Acids Res. 2011, 39(Web Server issue): W316-W322.
[16] Nastou KC, Tsaousis GN, Papandreou NC, et al. MBPpred: Proteome-wide detection of membrane lipid-binding proteins using profile Hidden Markov Models [J]. Biochim Biophys Acta. 2016, 1864(7):747-754.
[17] Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 [J]. Genome Biol. 2014, 15(12):550.
[18] 贺润丽, 王晓英, 韩毅丽, 等. 利用转录组分析款冬萜类化合物生物合成关键酶基因及表达特征[J]. 中草药, 2020, 51(20): 5302-5310.
[19] 刘丽君. 转录组分析鉴定连翘中控制连翘苷、连翘酯苷A含量及胚发育的候选基因[D]. 河南大学, 2020.
[20] Havaux M. Carotenoids as membrane stabilizers in chloroplasts[J]. Trends in Plant Science. 1998, 3(4): 147-151.