作者Jaeyun Sung在2020年和2024年的两篇文章,分别提出了计算个体肠道微生物健康指数的方法:
- A predictive index for health status using species-level gut microbiome profiling
- Gut Microbiome Wellness Index 2 enhances health status prediction from gut microbiome taxonomic profiles
研究设计
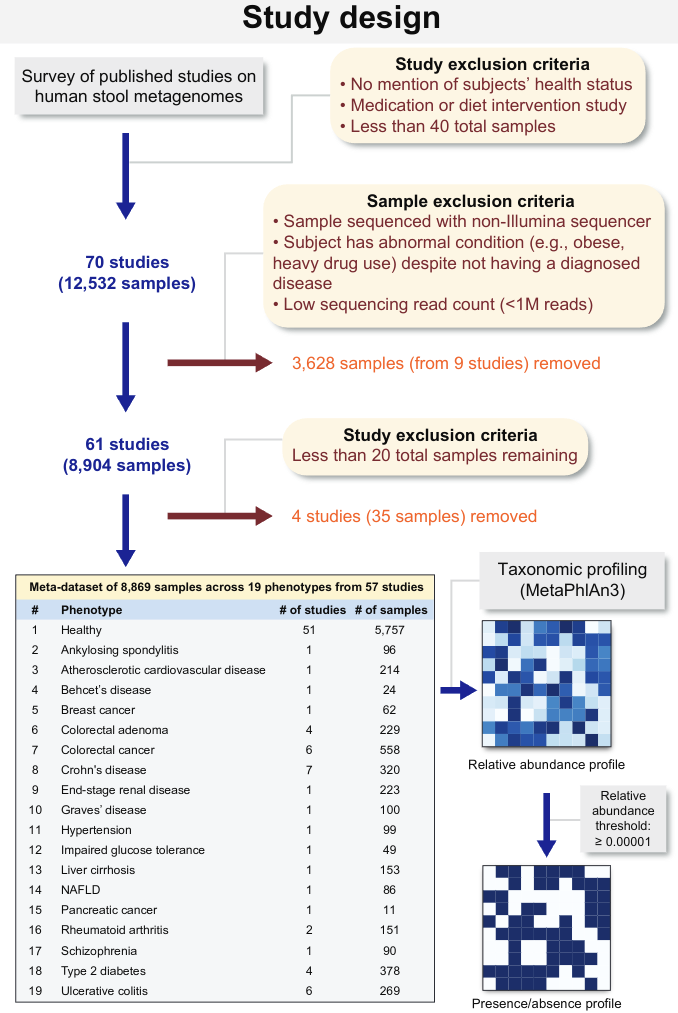
作者首先收集了70各个研究中12,532份肠道微生物样本,基于一些排除标准,最终有8,869份样本保留,其中57个健康的研究有5757份样本和18个非健康研究有3112份研究。所有样本采用相同的生物信息学流程处理。菌群丰度使用MetaPhlan3计算,根据相对丰度阈值>=0.00001确定每个样本的菌群存在/不存在。
Lasso惩罚逻辑回归提取健康和疾病相关分类群
使用Lasso惩罚逻辑回归(Lasso-penalized logistic regression)对菌群的矩阵进行分析,得到:
- 59个菌(taxa)的系数是正
- 3091个菌的系数是0
- 50个菌的系数是负
非零系数的菌包括,1个冈、3个目、7科、19属、79种
系数被定义为当样本中的相关分类群从缺失变为存在时,属于健康患者的样本的log-odds的期望变化
假设我们有一组数据,记录了不同年龄段的人群是否患病的情况。通过逻辑回归分析,我们得到了关于年龄这一自变量的Odds Ratio值,比如OR=1.1。
这个OR=1.1的含义是:在控制其他可能影响患病的因素(如性别、遗传、生活习惯等)不变的情况下,年龄每增加一岁,个体患病的概率与不患病的概率之比将增加10%。换句话说,如果一个人在某个年龄段患病的概率是某个值,那么当他年龄增加一岁时,他患病的“胜率”(即患病的概率与不患病的概率之比)将变为原来的1.1倍。
根据https://github.com/biobakery/MetaPhlAn/wiki/MetaPhlAn-3.1安装MetaPhlAn的数据库,作者使用MetaPhlAn的数据库的版本是mpa_v30_CHOCOPhlAn_201901,详情见。
下载指定版本的MetaPhlAn数据库到当前文件夹的db目录:
metaphlan --install --index mpa_v30_CHOCOPhlAn_201901 --bowtie2db db
docker 命令
docker run --rm -it \
-u $(id -u):$(id -g) \
-v $PWD:$PWD \
-w $PWD \
wybioinfo/gmwi2:1.6 \
metaphlan --install --index mpa_v30_CHOCOPhlAn_201901 --bowtie2db db
注意此时会比较慢,MetaPhlAn需要下载数据库并建立索引
Downloading MetaPhlAn database
Please note due to the size this might take a few minutes
Downloading http://cmprod1.cibio.unitn.it/biobakery3/metaphlan_databases/mpa_v30_CHOCOPhlAn_201901.tar
Downloading file of size: 366.62 MB
366.62 MB 100.00 % 16.09 MB/sec 0 min -0 sec
Downloading http://cmprod1.cibio.unitn.it/biobakery3/metaphlan_databases/mpa_v30_CHOCOPhlAn_201901.md5
Downloading file of size: 0.00 MB
0.01 MB 12800.00 % 31.60 MB/sec 0 min -0 sec
Decompressing db/mpa_v30_CHOCOPhlAn_201901.fna.bz2 into db/mpa_v30_CHOCOPhlAn_201901.fna
Building Bowtie2 indexes
from joblib import load
import numpy as np
import pandas as pd
df = pd.read_csv( "tiny_metaphlan.txt", sep="\t", skiprows=3, usecols=[0, 2], index_col=0).T
dummy_cols = list(set(gmwi2.feature_names_in_) - set(df.columns))
dummy_df = pd.DataFrame(np.zeros((1, len(dummy_cols))), columns=dummy_cols, index=["relative_abundance"])
df = pd.concat([dummy_df, df], axis=1)
df = df.copy()[["UNKNOWN"] + list(gmwi2.feature_names_in_)]
# normalize relative abundances
df = df.divide((100 - df["UNKNOWN"]), axis="rows")
df = df.drop(labels=["UNKNOWN"], axis=1)
presence_cutoff = 0.00001
gmwi2.decision_function(df > presence_cutoff)[0]
#mpa_v30_CHOCOPhlAn_201901
#/Users/daniel/opt/anaconda3/envs/test_gmwi2/bin/metaphlan SRR1761667_QC_1P.fastq.gz,SRR1761667_QC_2P.fastq.gz --index mpa_v30_CHOCOPhlAn_201901 --force --no_map --nproc 16 --input_type fastq -o SRR1761667_metaphlan.txt --add_viruses --unknown_estimation
#SampleID Metaphlan_Analysis
#clade_name NCBI_tax_id relative_abundance additional_species
UNKNOWN -1 70.84224
k__Bacteria 2 29.031894900524044
k__Archaea 2157 0.1258623750557678
k__Bacteria|p__Firmicutes 2|1239 11.989174109844733
df = pd.read_csv( "tiny_metaphlan.txt", sep="\t", skiprows=4, usecols=[0, 2], index_col=0).T
dummy_cols = list(set(gmwi2.feature_names_in_) - set(df.columns))
dummy_df = pd.DataFrame(np.zeros((1, len(dummy_cols))), columns=dummy_cols, index=["relative_abundance"])
df = pd.concat([dummy_df, df], axis=1)
df = df.copy()[list(gmwi2.feature_names_in_)]
# normalize relative abundances
# df = df.divide((100 - df["UNKNOWN"]), axis="rows")
# df = df.drop(labels=["UNKNOWN"], axis=1)
presence_cutoff = 0.00001
gmwi2.decision_function(df > presence_cutoff)[0]