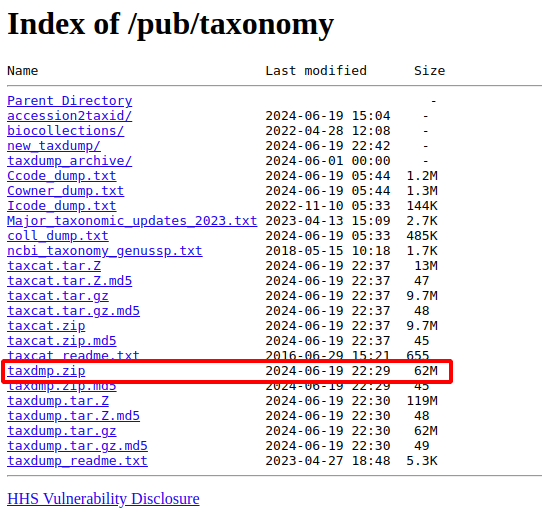
进入Taxonomy数据库: https://www.ncbi.nlm.nih.gov/taxonomy
library(tidyverse)
# tax_id -- the id of node associated with this name
# name_txt -- name itself
# unique name -- the unique variant of this name if name not unique
# name class -- (synonym, common name, ...)
names.0 <- read_delim("names.dmp",delim = "\t|\t",col_names=F) |>
mutate(X4=str_replace(X4,"\\t\\|","")) |>
select(tax_id=X1, name_txt=X2, unique_name=X3, name_class=X4)
head(names.0)
# nodes.dmp file consists of taxonomy nodes. The description for each node includes the following
# fields:
# tax_id -- node id in GenBank taxonomy database
# parent tax_id -- parent node id in GenBank taxonomy database
# rank -- rank of this node (superkingdom, kingdom, ...)
# embl code -- locus-name prefix; not unique
# division id -- see division.dmp file
# inherited div flag (1 or 0) -- 1 if node inherits division from parent
# genetic code id -- see gencode.dmp file
# inherited GC flag (1 or 0) -- 1 if node inherits genetic code from parent
# mitochondrial genetic code id -- see gencode.dmp file
# inherited MGC flag (1 or 0) -- 1 if node inherits mitochondrial gencode from parent
# GenBank hidden flag (1 or 0) -- 1 if name is suppressed in GenBank entry lineage
# hidden subtree root flag (1 or 0) -- 1 if this subtree has no sequence data yet
# comments -- free-text comments and citations
nodes.0 <- read_delim("nodes.dmp",delim = "\t|\t",col_names=F) |>
mutate(X13=str_replace(X13,"\\t\\|","")) |>
select(tax_id=X1, parent_tax_id=X2, rank=X3, embl_code=X4, division_id=X5,
inherited_div_flag=X6, genetic_code_id=X7,inherited_GC_flag=X8,
mitochondrial_genetic_code_id=X9,inherited_MGC_flag=X10,
GenBank_hidden_flag=X11,hidden_subtree_root_flag=X12,
comments=X13)
head(nodes.0)
unique(nodes.0$rank)
table(nodes.0$rank)
name_node <- nodes.0 |>
inner_join(names.0,by = "tax_id") |>
filter(name_class=="scientific name") |>
select(tax_id,rank_= rank,name=name_txt,name_txt,parent_tax_id)
library(RMySQL)
con <- dbConnect(RMySQL::MySQL(), dbname = "test", host = "192.168.10.177",
user = "root", password = "rootroot",port=23306)
# dbListTables(con)
# dbListFields(con, "taxonomy")
dbWriteTable(con, "taxonomy", name_node, overwrite=TRUE, row.names = FALSE)
superkingdom <- name_node |>
filter(rank_=='superkingdom')
kingdom <- name_node |>
filter(rank_=='kingdom')
phylum <- name_node |>
filter(parent_tax_id==2 & rank_=='phylum')
class <- name_node |>
filter(parent_tax_id %in% phylum$tax_id & rank_=='class')
order <- name_node |>
filter(parent_tax_id %in% class$tax_id & rank_=='order')
family <- name_node |>
filter(parent_tax_id %in% order$tax_id & rank_=='family')
genus <- name_node |>
filter(parent_tax_id %in% family$tax_id & rank_=='genus')
species <- name_node |>
filter(parent_tax_id %in% genus$tax_id & rank_=='species')
subspecies <- name_node |>
filter(parent_tax_id %in% species$tax_id & rank_=='subspecies')
bacteria <- Reduce(function(x,y){rbind(x,y)},list(superkingdom,kingdom,phylum,
class,order,family,genus,
species,subspecies),accumulate = F)
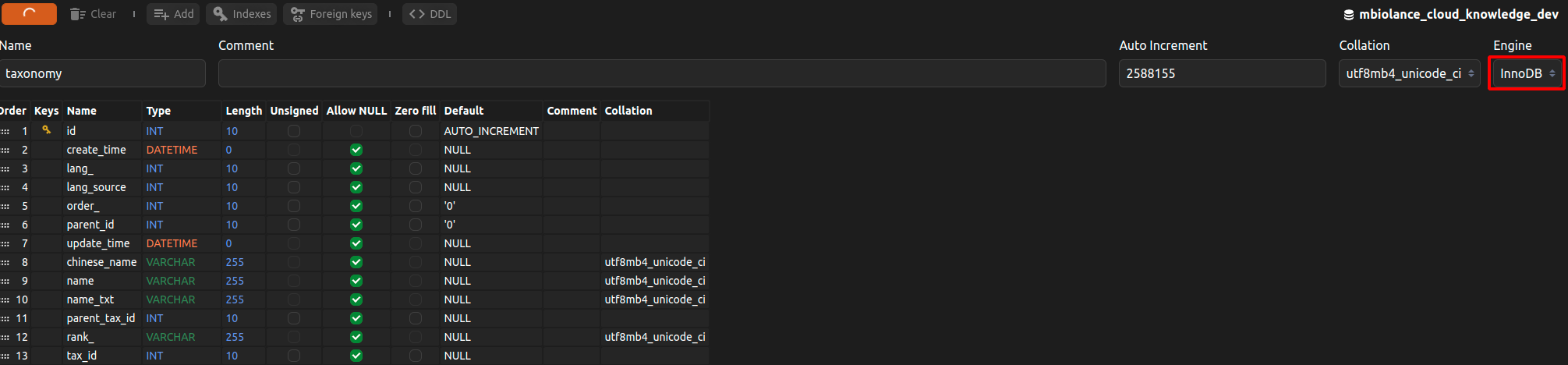
CREATE INDEX name_txt_index ON taxonomy (name_txt);
SHOW INDEX FROM taxonomy;
ALTER TABLE taxonomy DROP INDEX name_txt_index;
SELECT * FROM taxonomy where name_txt like '%Saccharomyces boulardii%'
SELECT * FROM taxonomy where name_txt like '%Weizmannia coagulans%'