Highlights
- 我们对 99,604 个常见的人类结构变异进行测序、解析和注释
- 55% 的 VNTR(variable number of tandem repeats) 定位于染色体末端,并与双链断裂相关
- 替代等位基因有助于通过短读长和新关联进行准确的基因分型
- 我们修补了参考基因组,并增加了开发泛人类基因组所需的多样性
Summary
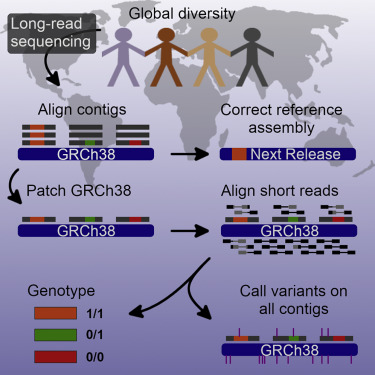
为了给人类结构变异(SVs)提供全面的资源,我们生成了长读长序列数据,并分析了15个人类基因组的SV。我们测序解析了 99,604 个插入(insertions)、缺失(deletions)和反转(inversions),包括 2,238 个 (1.6 Mbp),这些插入、缺失和反转在所有发现基因组中共享,另外 13,053 个 (6.9 Mbp) 占大多数,表明参考基因组中存在次要等位基因或错误。另外 440 个基因组的基因分型证实,独特的常染色质中最常见的 SV 现在已经序列解析。我们报告了对人类染色体最后 5 Mbp 的 9 倍 SV 偏差,其中近 55% 的 VNTR(可变数量的串联重复序列)映射到基因组的这一部分。我们确定了影响编码和非编码调控位点的 SV,从而改善了功能变异的注释和解释。这些数据为构建规范的人类参考提供了框架,并为开发能够捕获等位基因多样性的高级表示提供了资源。
Graphical Abstract
Introduction
目前的人类基因组参考(GRCh38)是由多个个体构建的,在任何给定的位点上,它都是单个人类单倍型的高质量表示。由于大部分参比来自大插入片段 BAC 克隆 (国际人类基因组测序联盟等,2001),来自单个克隆插入片段的序列偶然代表了这些位点的“人类参考”。近二十年来,这种序列一直是生物医学结果功能注释和解释的事实上的框架。虽然尚未完成,但一个版本的理想人类参考基因组将不包含任何空白,代表单一的人类单倍型,并在每个位点携带最常见的等位基因——本质上是一个规范的人类参考基因组(Schneider等人,2017).这尚未实现。参考基因组中的偏差和错误会影响序列读取比对的准确性和对人类遗传变异的正确解释(Brandt 等人,2015 年,Degner 等人,2009 年).
由于其对生物医学研究的重要性,人类基因组的质量自 2001 年首次发布以来一直在不断发展。精加工版本,GRCh35 (国际人类基因组测序联盟,2004 年),由来自八个个体的 2.85 Gbp 序列组成,并致力于针对剩余的差距。从那时起,许多错误已得到纠正,主组件已增长到 3.1 Gbp (Schneider等人,2017).最近的改进是通过采用诱饵序列和结合来自长读长测序技术的数据(Pendleton 等人,2015 年,Steinberg 等人,2014,Watson 等人,2013)以及来自单倍体水菌鼹鼠(Chaisson等人,2015a,Fan 等人,2002 年),这使我们能够更好地区分同源和等位基因变异。虽然目前的人类参考基因组构建GRCh38可以说是迄今为止构建的最完整的哺乳动物基因组参考,但其70%的序列仍然来自单个克隆文库RP11(Schneider等人,2017),它是从单个个体获得的,并在十多年前从大肠杆菌中繁殖的大插入 BAC 克隆组装而成。
由于发现类人猿物种之间和内部的广泛结构变异,单个人类参考基因组的组装特别复杂(Iafrate等人,2004,Locke 等人,2003 年,Sebat 等人,2004 年,Tuzun 等人,2005 年).广泛的基因组结构变异,现在在操作上定义为插入、重复、缺失和倒置>长度为 50 bp (Mills 等人,2011 年),意味着任何单一的人类单倍型都可能缺失或包含大多数人类中不存在的序列变异。因此,从单个单倍型在任何一个位置构建的人类基因组参考必须缺少遗传信息,或者携带我们物种不常见的罕见变异。
测序技术的最新进展现在使我们能够系统地对大段 (>10 kbp) 的天然 DNA 进行全基因组鸟枪法 (WGS) 测序,而无需在大肠杆菌中繁殖克隆插入片段。这对于结构变异特别有利,因为无论序列组成如何,长读长都为锚定和序列解析大多数结构变异 (SV) 提供了必要的上下文。以前,我们和其他人已经证明了长读长测序(Chaisson等人,2015a,Gordon 等人,2016 年,Pendleton 等人,2015 年,Seo 等人,2016,Shi 等人,2016),以提高SV检测的灵敏度。在这项研究中,我们以单倍体和人类基因组的多样性面板为目标,以创建最大的序列解析结构变异集合。该资源使我们能够发现人类物种共有的固定和主要等位基因SV,这些等位基因SV目前在人类参考基因组中缺失。这种主要等位基因SV的序列解析为开发经典人类参考基因组提供了重要的第一步。