- 酵母数据介绍
- 一、从Ensemble数据库下载参考基因组及注释文件
- 二、下载sra序列数据
- 三、将SRA转换成fastq文件
- 四、对测序数据进行质量评估
- 五、测序序列比对
- 六、计算基因短序列read count
- 七、DESeq2鉴定差异表达基因
- 从Ensembl获得基因注释信息
- Go和KEGG分析
- 参考
https://github.com/wangyang1749/yeast_data.git
酵母数据介绍
- 数据来自文章:In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse
- 该文章包括RAN-Seq数据、ChIP-Seq、DNA甲基化测序数据
- 背景:在酵母中没有DNA甲基化,该文章将DNA甲基化酶引入酵母。即转基因后,酵母基因组包含了基因甲基化,研究DNA甲基化影响表观遗传的状态。
- 本文介绍RAN-Seq数据的处理流程
一、从Ensemble数据库下载参考基因组及注释文件
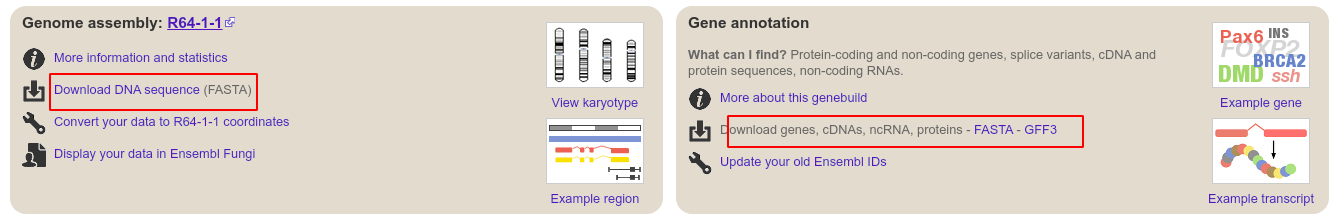
搜索 yeast genome ensemble,进入酵母的参考基因组网页,点击红色线框处。即可下载参考基因组和基因组注释文件
图片alt
使用以下命令下载,参考基因组和基因组注释文件
wget ftp://ftp.ensemblgenomes.org/pub/fungi/release-50/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.dna_rm.toplevel.fa.gz
wget ftp://ftp.ensemblgenomes.org/pub/release-50/fungi/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.50.gtf.gz
下载之后,使用gunzip解压。即可得到以下两个文件:
参考基因组

图片alt
基因组注释文件

图片alt
下载之后,可以使用sed查看染色体的数目:
sed -n '/^>/p' Saccharomyces_cerevisiae.R64-1-1.dna_rm.toplevel.fa
>I dna_rm:chromosome chromosome:R64-1-1:I:1:230218:1 REF
>II dna_rm:chromosome chromosome:R64-1-1:II:1:813184:1 REF
>III dna_rm:chromosome chromosome:R64-1-1:III:1:316620:1 REF
>IV dna_rm:chromosome chromosome:R64-1-1:IV:1:1531933:1 REF
>V dna_rm:chromosome chromosome:R64-1-1:V:1:576874:1 REF
>VI dna_rm:chromosome chromosome:R64-1-1:VI:1:270161:1 REF
>VII dna_rm:chromosome chromosome:R64-1-1:VII:1:1090940:1 REF
>VIII dna_rm:chromosome chromosome:R64-1-1:VIII:1:562643:1 REF
>IX dna_rm:chromosome chromosome:R64-1-1:IX:1:439888:1 REF
>X dna_rm:chromosome chromosome:R64-1-1:X:1:745751:1 REF
>XI dna_rm:chromosome chromosome:R64-1-1:XI:1:666816:1 REF
>XII dna_rm:chromosome chromosome:R64-1-1:XII:1:1078177:1 REF
>XIII dna_rm:chromosome chromosome:R64-1-1:XIII:1:924431:1 REF
>XIV dna_rm:chromosome chromosome:R64-1-1:XIV:1:784333:1 REF
>XV dna_rm:chromosome chromosome:R64-1-1:XV:1:1091291:1 REF
>XVI dna_rm:chromosome chromosome:R64-1-1:XVI:1:948066:1 REF
>Mito dna_rm:chromosome chromosome:R64-1-1:Mito:1:85779:1 REF
二、下载sra序列数据
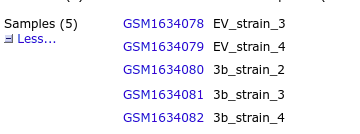
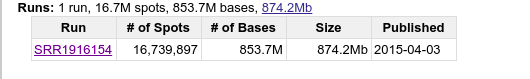
从GSE66911下载酵母的转录组测序数据文件

图片alt
图片alt
图片alt
使用如下命令下载:
wget -O SRR1916152.sra https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1916152/SRR1916152.1
wget -O SRR1916153.sra https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1916153/SRR1916153.1
wget -O SRR1916154.sra https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1916154/SRR1916154.1
wget -O SRR1916155.sra https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1916155/SRR1916155.1
wget -O SRR1916156.sra https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR1916156/SRR1916156.1
下载完成后,得到5个文件
| ctrl | treatment |
|---|---|
| EV_strain | 3d_strain |
| 2 | 3 |
三、将SRA转换成fastq文件
从ncbi下载得到是sra文件,需要使用SRA Toolit将sra文件转换为fastq文件
fastq-dump SRR1916152.sra
fastq-dump SRR1916153.sra
fastq-dump SRR1916154.sra
fastq-dump SRR1916155.sra
fastq-dump SRR1916156.sra
得到fastq文件是如下形式:

图片alt
shell 脚本可以方便的批量处理:
#!/bin/sh
for i in *sra
do
echo $i
fastq-dump --gzip --split-files $i
done
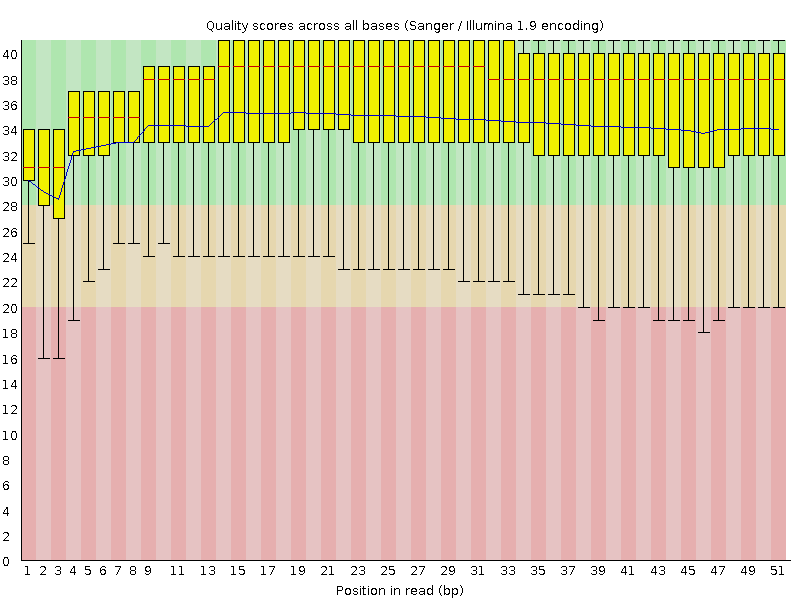
四、对测序数据进行质量评估
使用fastq对测序数据进行质量评估
命令如下:
fastqc SRR1916152.fastq
fastqc SRR1916153.fastq
fastqc SRR1916154.fastq
fastqc SRR1916155.fastq
fastqc SRR1916156.fastq
图片alt
五、测序序列比对
hisat2-build建立索引

图片alt
hisat2-build Saccharomyces_cerevisiae.R64-1-1.dna_rm.toplevel.fa yeast_ref
运行之后,产生如下文件:
图片alt
hisat2将read比对到参参考基因组
比对结果以sam格式文件保存
hisat2 -x ../../data/reference/yeast_ref -U ../../data/raw_data/SRR1916152.fastq > EV_str.sam

图片alt

图片alt
由于sam格式的文本文件过大,一般将其转换为bam格式的文件
samtools view -bS EV_str.sam -o EV_str.bam
对bam文件进行排序
samtools sort EV_str.bam -o EV_strain.sort.bam
一次分析所有的数据
hisat2 -x ../../data/reference/yeast_ref -U ../../data/raw_data/SRR1916152.fastq | samtools view -bS - | samtools sort - -o EV_3.bam
hisat2 -x ../../data/reference/yeast_ref -U ../../data/raw_data/SRR1916153.fastq | samtools view -bS - | samtools sort - -o EV_4.bam
hisat2 -x ../../data/reference/yeast_ref -U ../../data/raw_data/SRR1916154.fastq | samtools view -bS - | samtools sort - -o DNMT3B_2.bam
hisat2 -x ../../data/reference/yeast_ref -U ../../data/raw_data/SRR1916155.fastq | samtools view -bS - | samtools sort - -o DNMT3B_3.bam
hisat2 -x ../../data/reference/yeast_ref -U ../../data/raw_data/SRR1916156.fastq | samtools view -bS - | samtools sort - -o DNMT3B_4.bam
使用samtools tview查看比对信息
samtools tview EV_3.bam
建立bam文件的索引
samtools index EV_3.bam
统计比对信息
samtools flagstat EV_3.bam
igv比对信息可视化
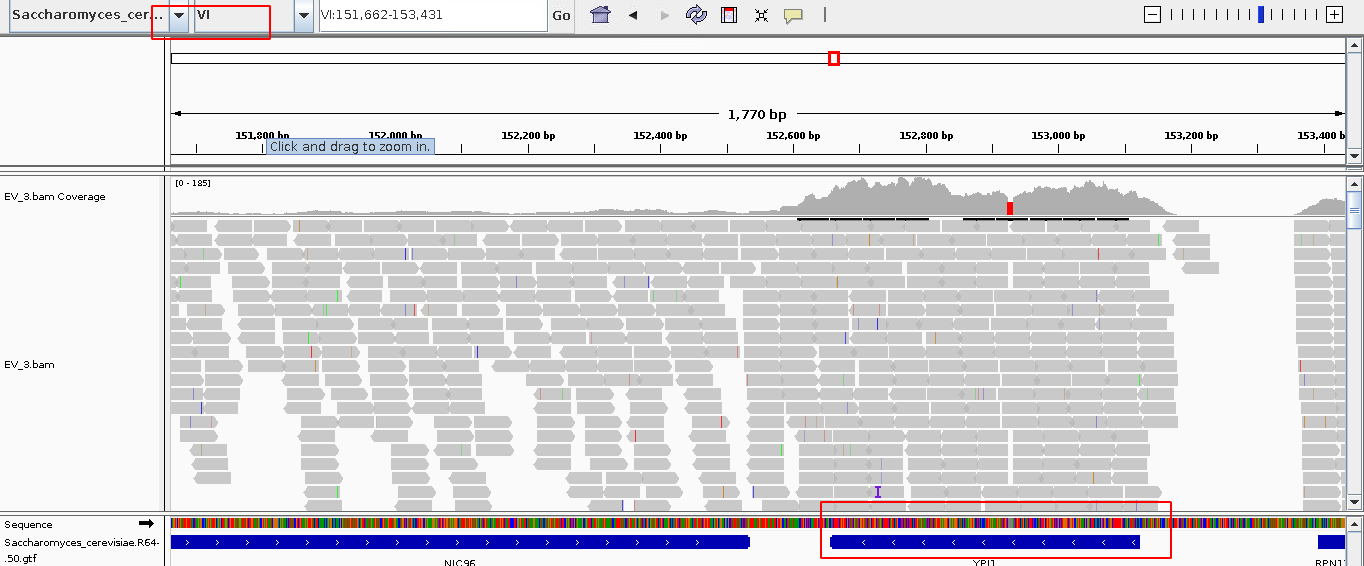
图片alt
六、计算基因短序列read count
安装HTSeq软件,计算每个基因中reads的数目
一次分析所有的数据
htseq-count -f bam -r pos ../read_aln/EV_3.bam ../../data/reference/Saccharomyces_cerevisiae.R64-1-1.48.gtf > EV_3.count.tab
htseq-count -f bam -r pos ../read_aln/EV_4.bam ../../data/reference/Saccharomyces_cerevisiae.R64-1-1.48.gtf > EV_4.count.tab
htseq-count -f bam -r pos ../read_aln/DNMT3B_2.bam ../../data/reference/Saccharomyces_cerevisiae.R64-1-1.48.gtf > DNMT3B_2.count.tab
htseq-count -f bam -r pos ../read_aln/DNMT3B_3.bam ../../data/reference/Saccharomyces_cerevisiae.R64-1-1.48.gtf > DNMT3B_3.count.tab
htseq-count -f bam -r pos ../read_aln/DNMT3B_4.bam ../../data/reference/Saccharomyces_cerevisiae.R64-1-1.48.gtf > DNMT3B_4.count.tab
七、DESeq2鉴定差异表达基因
PCA
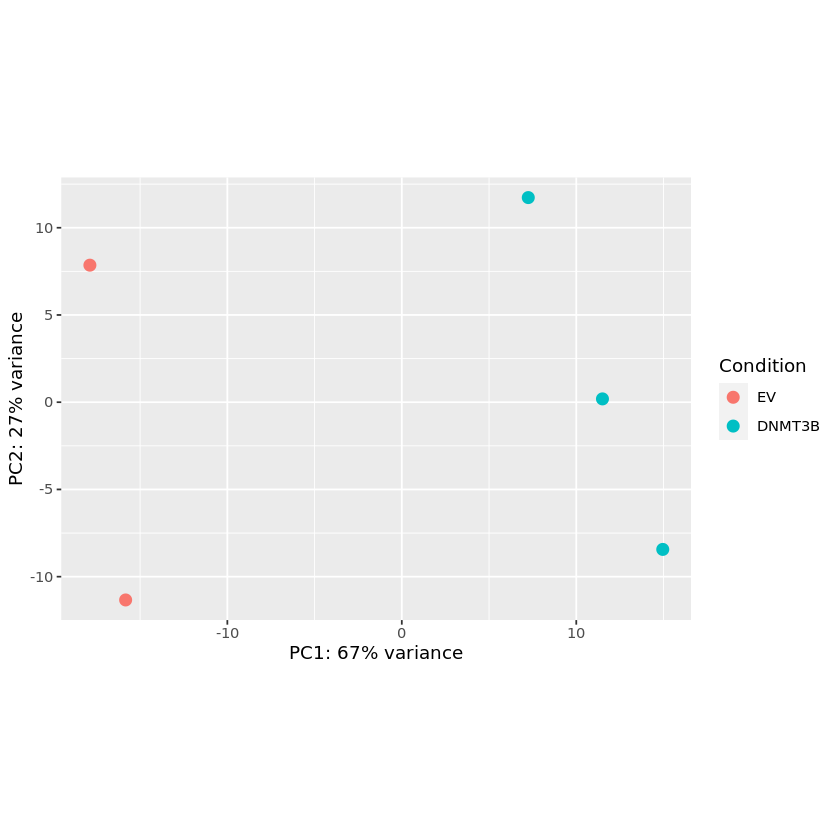
图片alt
MA-plot

图片alt
差异基因表

图片alt
- log2FoldChange:差异倍数取log
- padj:校准的pvalue
使用awk筛选基因
上调基因
awk '$7 < 0.05 && $3 > 2' yeast_DESeq2_DEG.txt | less -S
下调基因
awk '$7 < 0.05 && $3 < -2' yeast_DESeq2_DEG.txt | less -S
完整R代码如下:
setwd("~/workspace/jupyter/yeast_data/data/info/")
getwd()
count_tab <- read.table("result.csv",header=T)
rownames(count_tab) <- count_tab$gene_id # count_tab[,1]
# 去掉第一列
count_tab <- count_tab[,-c(1)]
# count_tab <- count_tab[c(1,3,4,2,5)] # 没用?
count_tab <- count_tab[c(1,4,2,3,5)]
head(count_tab)
colData <- read.table("AccGroup.csv",header=T)
colData$Condition <- factor(colData$Condition,c("EV","DNMT3B"))
colData
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = count_tab,
colData = colData,
design= ~ Condition)
#(nor_count <- counts(dds))
dds <- DESeq(dds)
# 将res转换为dataframe
resultsNames(dds) # lists the coefficients
res <- results(dds, name="Condition_DNMT3B_vs_EV")
res <- res[order(res$padj),]
resDF <- as.data.frame(res)
class(resDF)
#resDF
resDF$gene_id <- rownames(resDF) # 添加一列gene_id
resDF <- resDF[,c(7,1,2,3,4,5,6)] # 将gene_id放到第一列
head(resDF)
write.table(resDF,file="yeast_DESeq2_DEG.txt",sep="\t",quote=F,row.names=F)
# or to shrink log fold changes association with condition:
# res <- lfcShrink(dds, coef="condition_trt_vs_untrt", type="apeglm")
vsd <- vst(dds,blind=FALSE) # 数据均一化
vsd
library('ggplot2')
pcaData <- plotPCA(vsd, intgroup=c("Condition"), returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
plot_fig <- ggplot(pcaData, aes(PC1, PC2, color=Condition)) +
geom_point(size=3) +
xlab(paste0("PC1: ",percentVar[1],"% variance")) +
ylab(paste0("PC2: ",percentVar[2],"% variance")) +
coord_fixed()
# 保存图片
ggsave(plot_fig,filename = "yeast_DESeq2_PCA.pdf")
plot_fig
# pdf("yesat_DESeq2_MAplot.pdf")
test <- plotMA(res, ylim=c(-2,2))
# dev.off()
从Ensembl获得基因注释信息
图片alt
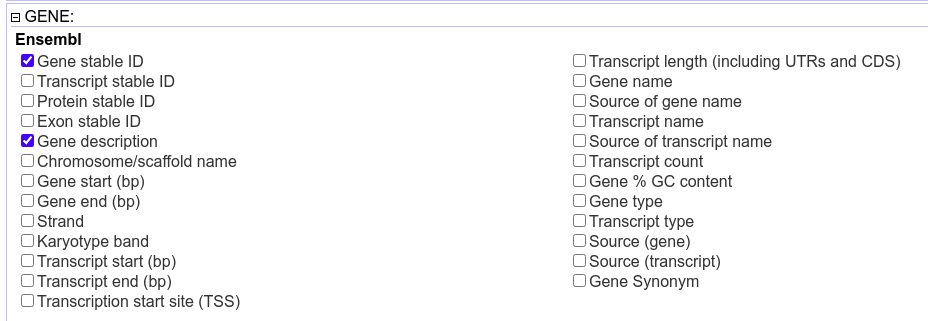
图片alt

图片alt
有些物种没有对应的注释信息
Go和KEGG分析
筛选获得差异基因
awk '$7 < 0.05 ' yeast_DESeq2_DEG.txt | cut -f1 > yeast_DESeq2_DEG.list
使用在线软件David进行富集分析:
KEGG
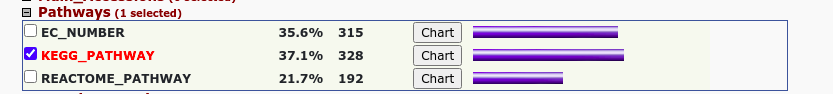
图片alt

图片alt

图片alt
GO
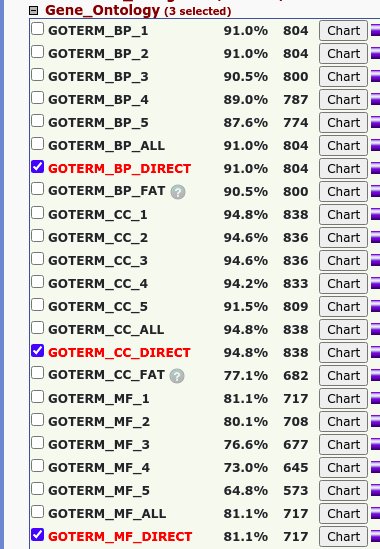
图片alt

图片alt

图片alt