A review of methods and databases for metagenomic classification and assembly
Abstract
在过去的十年里,微生物组研究迅速发展,新方法层出不穷,试图理解庞大而复杂的数据集。在这里,我们调查了分析微生物组数据的两种主要方法:read classification和metagenomic assembly,并回顾了这些方法面临的一些挑战。所有的方法都依赖于公共基因组数据库,我们还讨论了这些数据库的内容,以及它们的质量如何直接影响我们解释微生物组样本的能力。
Introduction
由于基因组测序效率的显著提高,微生物组研究一直在迅速扩展。随着实验的多样性和复杂性的增长,用于分析这些实验的方法和数据库也在增长。越来越大的数据集给计算方法带来了越来越多的挑战,这些方法必须最大限度地减少处理和内存需求,以提供快速周转,并避免使大多数研究实验室可用的计算资源不堪重负。基因组数量和种类的快速增加也带来了许多挑战,部分原因是将传统的分类命名方案融入微生物世界所需的努力,我们现在知道,微生物世界比科学家在遥远的过去首次创建分类命名方案时所意识到的要丰富和复杂得多。“草案”基因组测序的快速步伐带来了额外的挑战,它已经产生了数以万计的新基因组,其中许多是高度分散和不完整的。正如我们下面所讨论的,如果在没有仔细审查的情况下使用这些基因组,这些基因组的可变质量可能会导致意想不到的错误结果
这篇综述讨论了分析宏基因组学数据的计算挑战,重点是方法,但也包括对微生物分类学和基因组资源的讨论,尽管它们至关重要,但在基准研究和工具综述中很少讨论。我们首先回顾了术语,并对marker gene sequencing、shotgun metagenome sequencing和meta-transcriptome sequencing进行了比较,所有这些有时都包含在metagenomics一词中。
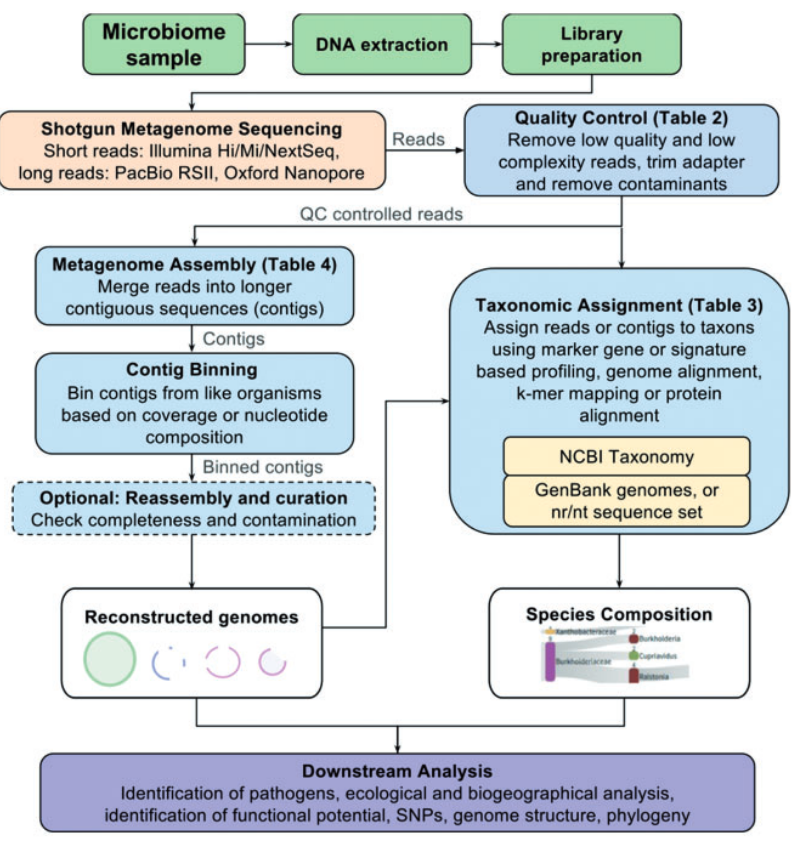
宏基因组学数据的通用分析程序。请注意,一些分析步骤的顺序可以打乱。例如,读取可能在assembly之前或分类分配之前进行装箱,因此下游算法只能处理数据的子集。
Metagenomic analysis
在质量控制之后,读数可以被组装成称为重叠群的更长的连续序列,也可以直接传递给分类分类器。每个读数的分类是一种分仓形式,因为它将读数分组到与其分类单元ID相对应的分仓中。分仓也可以使用其他属性来完成,如组成和共丰度谱,尽管这些方法通常需要将读数组装成更长的重叠群,这为分析提供了更好的统计数据。当分析只返回不同分类群的估计丰度(而不是每个读数的分类)时,我们称其为分类分析。选择基于组装的分析与直接的reads分类取决于研究问题。
直接分类学分类有助于定量群落分析和鉴定数据库中具有近亲的生物。与基于标记基因的群落分析相比,宏基因组鸟枪测序减轻了引物选择的偏见,并能够检测生命所有领域的生物体,假设DNA可以从目标环境中提取。研究人员可以使用生态和生物地理学措施来量化微生物群落的结构,如群落的物种多样性、丰富度和一致性。在临床微生物学中,重点通常是是否存在传染性病原体,可以通过将读数与参考数据库进行匹配来识别。尽管参考数据库中有许多完整的基因组,对人类相关微生物的研究相对较好,但一些病原体仍然没有测序,而另一些病原体是最近才使用宏基因组测序发现的。通过将reads与途径或基因数据库相匹配,可以深入了解微生物组的功能潜力。
当数据库中没有物种的近亲时,就像来自未探索生态位的样本经常发生的那样,对读数进行组装和装箱可能是分析中有用的第一步。对装箱的基因组草案的分析可以更定性地了解未培养微生物的生理学。通过鉴定重叠群箱中的单拷贝和保守基因,可以评估分类学、基因组完整性以及污染。宏基因组(草案)组装的一些最新发现包括鉴定Smithella spp.用于石油和石蜡降解的酶[57-59],以及深入了解甲烷生物反应器中的代谢途径和微生物之间的相互作用。
Metagenomic classification
宏基因组分类工具将序列与微生物基因组数据库进行匹配,以识别每个序列的分类单元。在宏基因组学的早期,最好的策略是使用BLAST将每个读数与GenBank中的所有序列进行比较。随着参考数据库和测序数据集的规模不断扩大,使用BLAST进行比对在计算上变得不可行,这导致了宏基因组学分类器的发展,该分类器提供了更快的结果,尽管通常比BLAST灵敏度更低。有些程序返回每次读取的赋值,而另一些程序只提供样本的整体组成。匹配步骤使用了多种策略:aligning reads, mapping k-mers, using complete genomes, aligning marker genes only or translating the DNA and aligning to protein sequences。最近的研究试图根据准确性和速度来衡量宏基因组学分类器的性能,尽管这些研究受到对模拟数据(不可避免)依赖的限制。
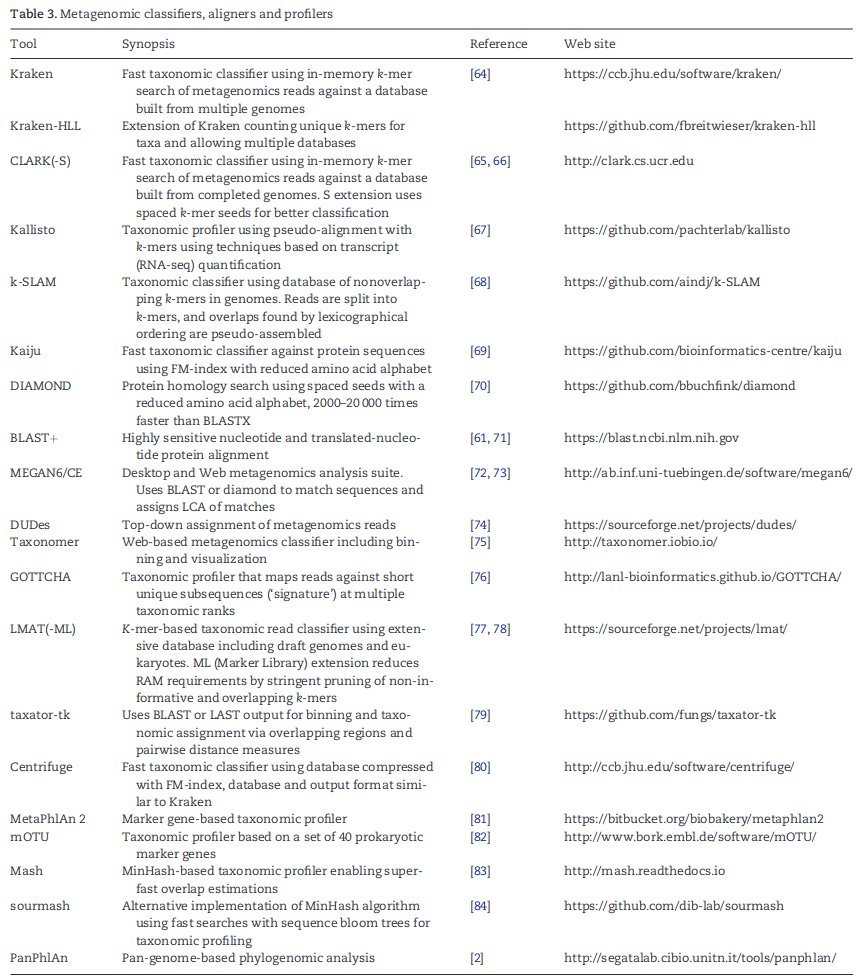
Metagenomic classifiers, aligners and profilers
Taxonomic profiling with marker gene-based and other approaches
标记基因方法识别一组分支特异性的单拷贝基因,因此这些基因中的一个的识别可以用作相关分支成员存在的证据。这允许更快的分配,因为即使有一百万或更多的基因(如MetaPhlAn[81]),该数据库也远小于包含所有物种完整基因组的数据库。然后可以使用快速、灵敏的对准器进行分配,例如MetaPhlAn使用的Bowtie2和Phylsifit和mOTU使用的HMMER。GOTTCHA基于唯一的24个碱基对片段生成具有唯一基因组特征的数据库,并用bwa-mem对其进行索引。GOTTCHA可以输出二元分类(存在/不存在调用)或基于基因组特征覆盖率的分类图谱。单拷贝标记基因的使用原则上应该使丰度估计更加精确,尽管不可能知道基因组不完整物种的基因拷贝数。因为标记基因方法在每个基因组中只识别少数基因,所以样本中的大多数读数根本没有得到分类;相反,这些算法提供了微生物组成,以它们在样本中识别的所有分类群的相对丰度表示。
宏基因组学分析的另一种方法是使用在Mash和sourmash中实现的MinHash签名的重叠。MinHashes允许人们非常有效地估计数据集的相似性,例如GenBank中所有微生物基因组和宏基因组学数据集之间的重叠。MinHash搜索数据库体积小,构建和搜索速度快,可以在笔记本电脑上搜索整个GenBank数据库。
Nucleotide taxonomic classification and quantification
Kraken是第一种快速鉴定宏基因组样本中所有读数的方法。它使用一种依赖于精确k-mer匹配的算法来实现这一点,用简单的表查找取代对齐(这需要更多的计算工作)。Kraken构建了一个数据库,该数据库与每个基因组中的每个k-mer一起存储该k-mer的物种标识符(分类ID)。当在两个或多个分类群中发现k-mer时,Kraken将这些分类群的最低共同祖先(LCA)与该k-mer存储在一起。数据库k-mer及其分类群保存在压缩的查找表中,该查找表可以快速查询与宏基因组学数据集的读数(或重叠群)中发现的k-mer的精确匹配。CLARK使用类似的方法,建立物种或属级特定k-mer的数据库,并放弃任何映射到更高级别的k-mer。Kraken和CLARK都默认设置k =31,尽管数据库可以用任何长度的k-mer构建。k的选择反映了敏感性和特异性之间的重要权衡:过长的k-mer可能由于测序错误或物种和菌株之间的真正差异而无法匹配,而过短的k-mer将产生与许多基因组的非特异性(和错误)匹配。使用固定k-mers的另一种方法是间隔或自适应(可变长度)种子,它对只有一个子集碱基必须完全匹配的模式进行编码。Kraken使用间隔种子的扩展在科和属级别的分类中显示出更好的准确性,但在物种级别的分类精度较低。针对CLARK开发了类似的扩展。请注意,Kraken 映射 reads的是分类树,而不是物种或属等特定级别。Bracken[94]是Kraken的扩展,它基于贝叶斯概率算法估计物种或属水平的丰度。Livermore宏基因组学分析工具包(LMAT)是基于k-mer的分类器,使用比Kraken和CLARK更小的默认k-mer大小(k = 20)