Kraken: ultrafast metagenomic sequence classification using exact alignments
Abstract
Kraken是一个为宏基因组DNA序列分配taxonomic labels的超快且高度准确的程序。以前为这项任务设计的程序相对缓慢,计算成本高昂,迫使研究人员使用更快的丰度估计程序,这些程序只对宏基因组数据的子集进行分类。使用k-mers的精确比对,Kraken实现了与最快BLAST程序相当的分类精度。在最快的模式下,Kraken以每分钟超过410万次的速度对100个碱基对读数进行分类,比Megablast快909倍,比丰度估计程序MetaPhlAn快11倍。Kraken可在http://ccb.jhu.edu/software/kraken/。
Background
宏基因组学是对直接从环境中获得的基因组序列的研究,在过去十年中已成为一个越来越受欢迎的研究领域。在研究海水、酸性矿井排水和人体等多种环境的项目中,宏基因组学使研究人员能够在不需要分离和培养单个微生物的情况下创建环境微生物生命的图片。结合快速测序DNA的能力,宏基因组学项目可以生成大量的序列数据来描述这些以前看不见的世界。
对于许多宏基因组样本,在测序时,样本中存在的物种、属甚至门在很大程度上是未知的,测序的目标是尽可能精确地确定这种微生物组成。当然,如果一个生物体与以前看到的任何生物都完全不同,那么除了将其标记为新颖之外,就无法对其DNA序列进行表征。然而,许多物种与已知物种有一些可检测的相似性,这种相似性可以通过敏感的比对算法来检测。最著名的这种算法,也是为未知序列分配分类标签的最佳方法之一,是BLAST程序,它可以通过找到与大型基因组序列数据库的最佳比对来对序列进行分类。尽管BLAST不是为宏基因组序列设计的,但它很容易适应这个问题,并且它仍然是可用的最佳方法之一。
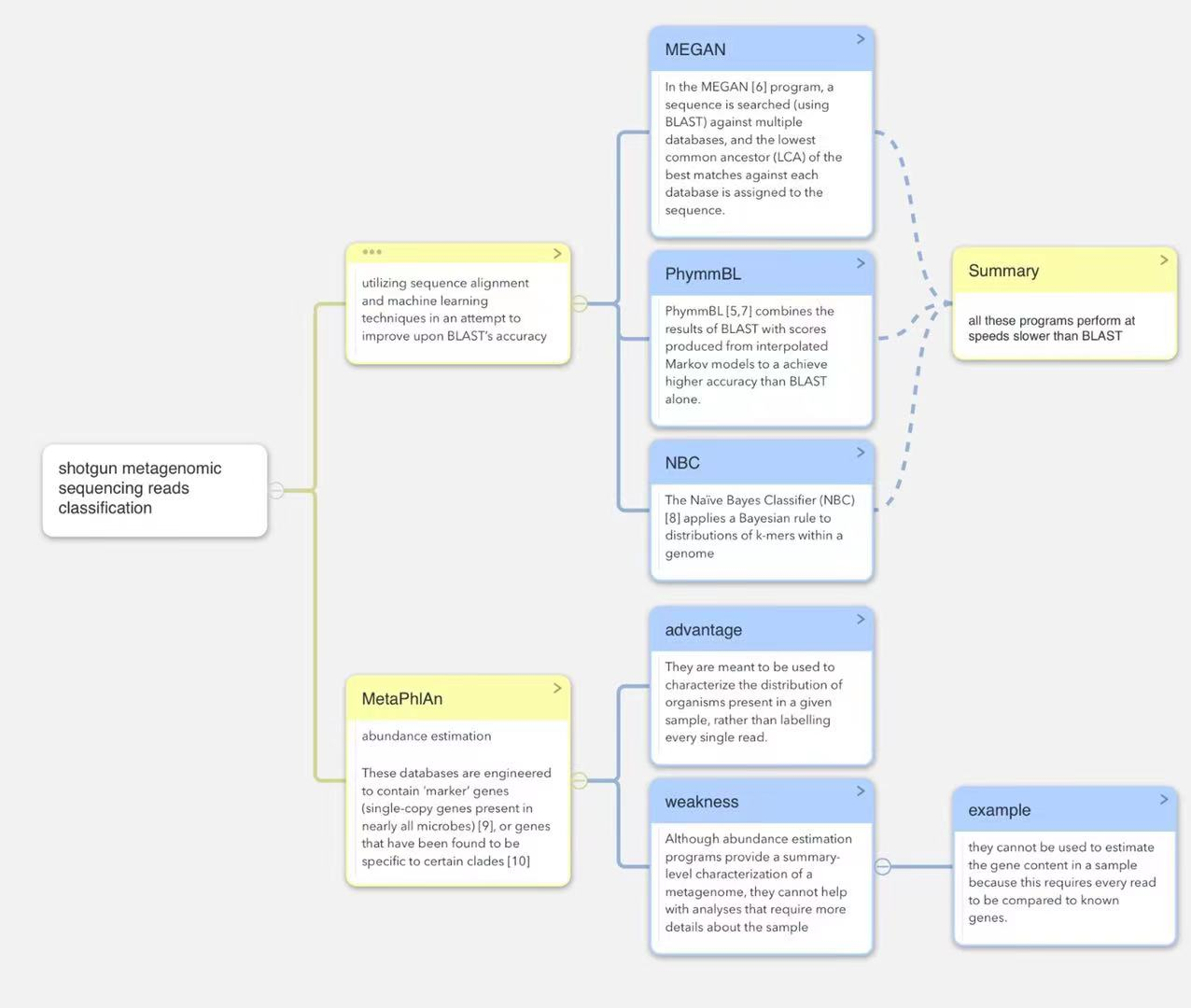
已经提出了利用序列比对和机器学习技术的其他序列分类方法,试图提高BLAST的准确性。在MEGAN程序中,针对多个数据库搜索序列(使用BLAST),并将针对每个数据库的最佳匹配的最低公共祖先(lowest common ancestor, LCA)分配给该序列。PhymmBL将BLAST的结果与插值马尔可夫模型产生的分数相结合,以实现比单独BLAST更高的精度。朴素贝叶斯分类器(NBC)将贝叶斯规则应用于基因组内k-mers的分布。然而,所有这些程序的执行速度都比BLAST慢,BLAST本身需要大量的CPU时间来对齐典型的Illumina测序运行产生的数百万个序列。这种处理负担要求如此之高,以至于它提出了另一种更快的宏基因组序列分析方法:丰度估计。
丰度估计程序的工作原理是创建一个比所有基因组的集合小得多的数据库,这使他们能够进行分类比试图识别数据集中的每次读取的方法快得多。这些数据库被设计为包含“标记”基因(几乎所有微生物中都存在单拷贝基因),或被发现对某些分支具有特异性的基因。由于数据库只包含每个基因组的很小样本,这些程序只能对典型宏基因组学样本中的一小部分序列进行分类。它们旨在用于表征给定样本中存在的生物体的分布,而不是标记每一个读数。例如,人类微生物组项目的初步分析使用了其中一个程序MetaPhlAn来分析从数百人中收集的数万亿个碱基(terababs)的宏基因组序列。尽管丰度估计程序提供了宏基因组的总结级表征,但它们无法帮助进行需要更多样本细节的分析。例如,它们不能用于估计样本中的基因含量,因为这需要将每个读数与已知基因进行比较。如果一个样本包含来自一个物种的大量读数,那么有时可以组装这些读数来重建部分或全部基因组,然后对产生的重叠群进行分类。
在这里,我们描述了Kraken,这是一种新的序列分类工具,其准确性与最佳序列分类技术相当,其速度远远超过分类器和丰度估计程序。这种速度优势在很大程度上源于使用k-mers的精确匹配数据库查询,而不是序列的不精确比对。其准确性是由于测序的微生物基因组数量非常庞大且仍在增长,目前已超过8500个,这使得以前很可能见过来自特定物种的非常相似的序列。通过使用一种新的算法来处理其数据库返回的不同结果,Kraken能够实现属级灵敏度和精度,与最快的BLAST程序Megablast获得的灵敏度和精度非常相似。