A practical guide to amplicon and metagenomic analysis of microbiome data
ABSTRACT
高通量测序(HTS)的进展促进了微生物组研究领域的快速发展,目前正在生成大量的微生物组数据集。然而,软件工具的多样性和分析管道的复杂性使得进入该领域变得困难。在这里,我们系统地总结了微生物组方法的优点和局限性。然后,我们推荐用于扩增子和宏基因组分析的特定管道,并描述常用的软件和数据库,以帮助研究人员选择合适的工具。此外,我们介绍了适用于微生物组分析的统计和可视化方法,包括α和β多样性、分类组成、差异比较、相关性、网络、机器学习、进化、来源追踪和常见的可视化风格,以帮助研究人员做出明智的选择。最后,介绍了一个逐步可重复的分析指南。我们希望这篇综述将使研究人员能够更有效地进行数据分析,并快速选择合适的工具,以便有效地挖掘数据背后的生物学意义。
INTRODUCTION
微生物组是指整个微生物栖息地,包括其微生物、基因组和周围环。随着高通量测序(HTS)技术和数据分析方法的发展,微生物组在人类、动物、植物和环境中的作用近年来逐渐清晰。这些发现彻底改变了我们对微生物组的理解。一些国家已经启动了成功的国际微生物组项目,如NIH Human Microbiome Project(HMP)、Metagenomics of the Human Intestinal Tract(MetaHIT)、integrative HMP(iHMP)和Chinese Academy of Sciences Initiative of Microbiome(CAS-CMI)。这些项目取得了令人瞩目的成就,将微生物组研究推向了黄金时代。
扩增子和宏基因组分析的框架是在过去十年中建立的,然而微生物的分析方法和标准在过去几年中发展迅速。例如,在基于标记基因的扩增子数据分析中,有人提议用amplicon sequence variants(ASV)取代operational taxonomic units(OTU)(2016, DADA2)。最近发布了下一代微生物组分析管道QIME2(2019),这是一个可复制、交互式、高效、社区支持的平台。此外,最近还提出了分类分类的新方法、机器学习和多组学综合分析。
HTS和分析方法的发展为微生物组的结构和功能提供了新的见解。然而,这些新的发展使研究人员,特别是那些没有生物信息学背景的研究人员,很难选择合适的软件和管道。在这篇综述中,我们讨论了广泛使用的微生物组分析软件包,总结了它们的优势和局限性,并为选择和使用这些工具提供了样本代码和建议。
HTS METHODS OF MICROBIOME ANALYSIS
微生物组研究的第一步是了解特定HTS方法的优势和局限性。这些方法主要用于三种类型的分析:微生物、DNA和mRNA水平分析(图1A)。应根据样本类型和研究目标选择适当的方法。
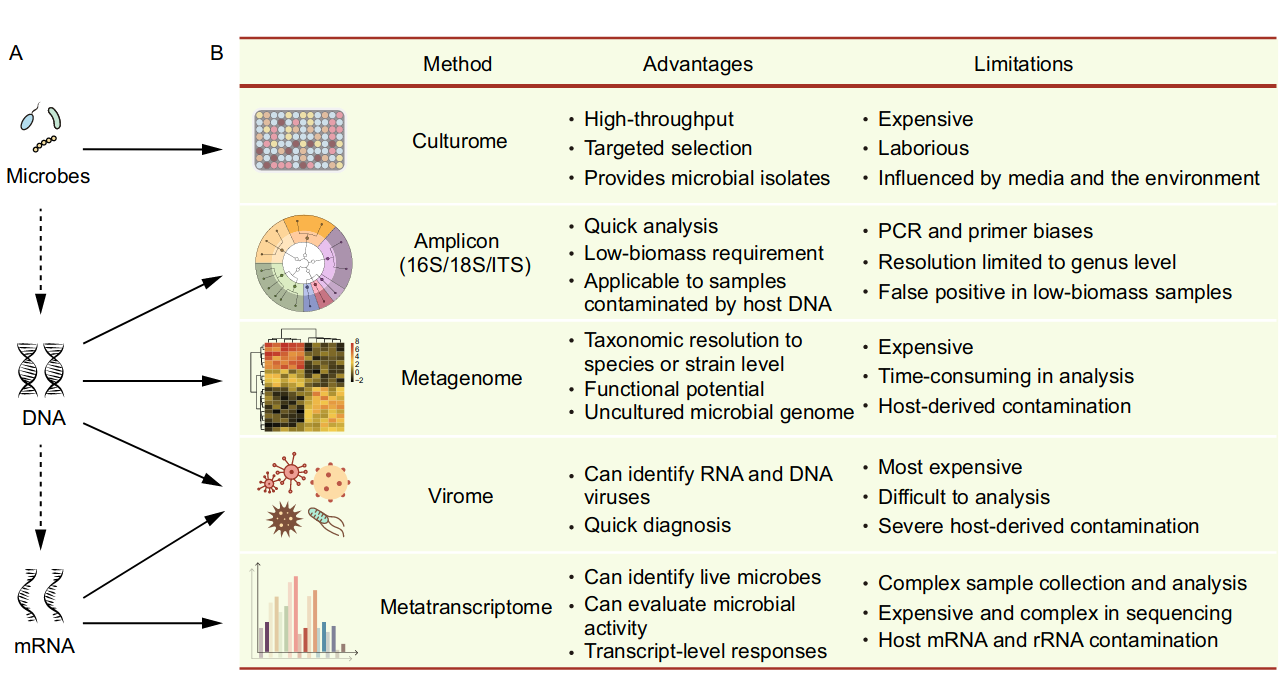
图1。HTS方法在微生物组研究中的优势和局限性。用于不同分析级别的HTS方法介绍。在分子水平上,微生物组研究分为三种类型:微生物、DNA和信使核糖核酸。相应的研究技术包括培养组、扩增子、宏基因组、宏病毒组和宏转录组分析。B各种HTS方法用于微生物组分析的优点和局限性。
Culturome是一种在微生物水平上培养和鉴定微生物的高通量方法(图1A)。微生物分离物如下获得。首先,将样品压碎,在液体培养基中凭经验稀释,并分布在96孔微量滴定板或培养皿中。第二,将平板在室温下培养20天。第三,对每个孔中的微生物进行扩增子测序,并选择具有纯非冗余菌落的孔作为候选。第四,对候选物进行纯化并进行16S rDNA全长Sanger测序。最后,保存了新鉴定的纯分离株。Culturome是获得细菌库存的最有效方法,但价格昂贵且劳动密集(图1B)。该方法已用于人类的微生物组分析、小鼠、海洋沉积物、拟南芥和水稻。这些研究不仅扩展了宏基因组分析的分类和功能数据库目录,还为实验验证提供了细菌储备。
DNA易于提取、保存和测序,这使研究人员能够开发各种HTS方法。微生物组常用的HTS方法是扩增子和宏基因组测序。扩增子测序是用于微生物组分析的最广泛使用的HTS方法,几乎可以应用于所有样本类型。扩增子测序中使用的主要标记基因包括原核生物的16S核糖体DNA(rDNA)和真核生物的18S rDNA和内部转录间隔区(ITS)。16S rDNA扩增子测序是最常用的方法,但目前可用的引物阵列令人困惑。选择引物的一个好方法是基于SILVA数据库和宿主因素(包括叶绿体、线粒体、核糖体和其他潜在的非特异性扩增来源),使用真实样本或电子PCR来评估其特异性和总覆盖率。或者,研究人员可以参考已发表研究中使用的与他们自己相似的引物,这将节省方法优化的时间,并有助于比较研究之间的结果。两步PCR通常用于扩增,并在文库制备过程中向每个样本添加条形码和适配器。样本测序通常在Illumina MiSeq、HiSeq 2500或NovaSeq 6000平台上以配对末端250碱基(PE250)模式进行,每个样本产生50000–100000个读数。扩增子测序可以应用于低生物量样本或被宿主DNA污染的样本。然而,这种技术只能达到属级别的分辨率。此外,它对所选择的特异性引物和PCR循环数敏感,这可能导致下游分析中的一些假阳性或假阴性结果(图1B)。
宏基因组测序比扩增子测序提供了更多的信息,但使用这种技术的成本更高。对于人类粪便等“纯”样本,在宏基因组项目中,每个样本可接受的测序数据量在6至9千兆字节(GB)之间。文库建设和测序的相应价格从100美元到300美元不等。对于含有复杂微生物群或被宿主来源的DNA污染的样本,每个样本所需的测序输出范围为30至300 GB(Xu et al.,2018)。简言之,16S rDNA扩增子测序可用于研究细菌和/或古菌组成。如果需要更高的分类分辨率和功能信息,则建议对宏基因组测序进行进一步分析。当然,假设有足够的项目资金,宏基因组测序可以直接用于样本量较小的研究。
宏转录组测序可以分析微生物群落中的mRNA,量化基因表达水平,并为原位微生物群落的功能探索提供快照。值得注意的是,为了获得微生物群的转录信息,应该去除宿主RNA和其他rRNA。
由于病毒的遗传物质是DNA或RNA,从技术上讲,宏病毒组研究涉及宏基因组和宏转录组分析的结合(图1A和1B)。由于样本中病毒的生物量较低,病毒富集或去除宿主DNA是获得足够数量的病毒DNA或RNA进行分析的必要步骤(图1B)。
测序方法的选择取决于科学问题和样本类型。整合不同的方法是可取的,因为多组学可以深入了解微生物组的分类和功能。在实践中,由于时间和成本的限制,大多数研究人员只选择一种或两种HTS方法进行分析。尽管扩增子测序只能提供微生物群的分类组成,但它具有成本效益(每个样本20–50美元),可以应用于大规模研究。此外,扩增子测序产生的数据量相对较小,并且分析快速且易于执行。例如,使用普通笔记本电脑可以在一天内完成100个扩增子样本的数据分析。因此,扩增子测序经常被用于开拓性研究。与扩增子测序相反,宏基因组测序不仅将分类学分辨率扩展到物种或菌株水平,还提供了潜在的功能信息。宏基因组测序也使得从短读数中组装微生物基因组成为可能。然而,它对低生物量样本或被宿主基因组严重污染的样本表现不佳。
ANALYSIS PIPELINES
“分析管道”是指一个特定的程序或脚本,它将几个甚至几十个软件程序按一定的顺序有机地组合在一起,以完成复杂的分析任务。截至2020年1月23日,“扩增子”和“宏基因组”在谷歌学者中的提及次数分别超过20万次和4万次。由于它们的广泛使用,我们将讨论目前扩增子和宏基因组分析的最佳实践管道。研究人员应该熟悉Shell环境和R语言,我们在之前的综述中对此进行了讨论。
Amplicon analysis
扩增子分析的第一阶段是将原始读数(通常为fastq格式)转换为特征表(图2A)。原始读数通常为成对端250碱基(PE250)模式,并由Illumina平台生成。其他平台,包括Ion Torrent、PacBio和Nanopore,在本综述中没有讨论,可能不适用于下面讨论的分析管道。首先,基于它们的条形码序列对原始扩增子配对末端读数进行分组(解复用)。然后将成对的读数合并以获得扩增子序列,并去除条形码和引物。通常需要质量控制步骤来去除低质量的扩增子序列。通常需要质量控制步骤来去除低质量的扩增子序列。所有这些步骤都可以使用USEARCH或QIIME完成。或者,测序服务提供商提供的干净的扩增子数据可以用于下一次分析。
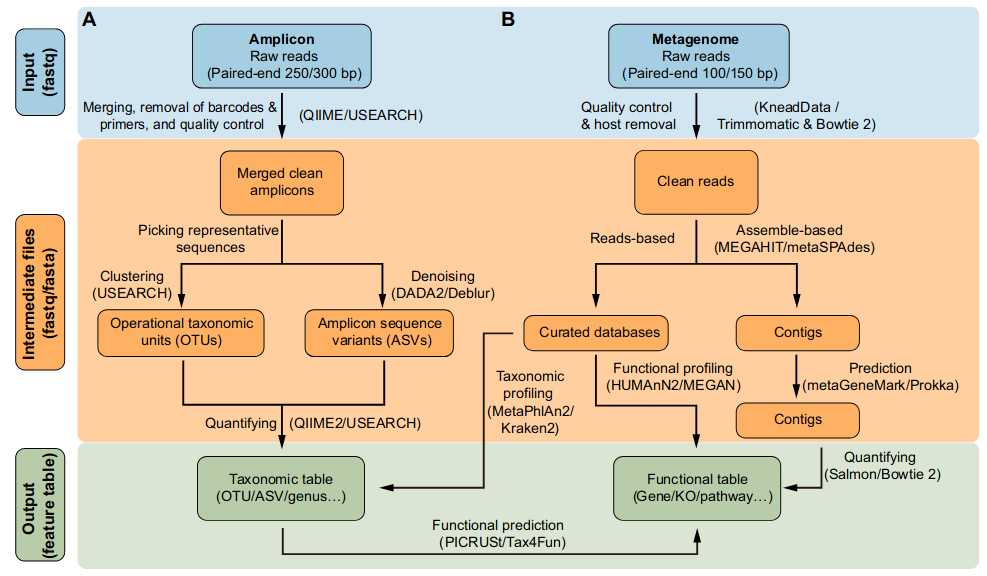
选择具有代表性的序列作为物种的代理是扩增子分析的关键步骤。代表性序列选择的两种主要方法是对OTU进行聚类和对ASV进行去噪。UPARSE算法将具有97%相似性的序列聚类为OTU。然而,这种方法可能无法检测到物种或菌株之间的细微差异。DADA2是最近开发的一种去噪算法,它将ASV输出为更精确的代表序列。该去噪方法可用于DADA2的成对/单个去噪、QIME 2中的Deblur的16S去噪和USEARCH中的-unise3去噪。最后,可以通过量化每个样本中特征序列的频率来获得特征表(OTU/ASV表)。同时,特征序列可以被划分为分类学,通常在界、门、纲、目、科、属和物种级别,提供了微生物群的降维视角。
通常,16S rDNA扩增子测序只能用于获得有关分类学组成的信息。然而,已经开发了许多可用的软件包来预测潜在的功能信息。这一预测背后的原理是将16S rDNA序列或分类学信息与文献中的功能描述联系起来。PICRUSt基于Greengenes数据库的OTU表,可用于预测Kyoto Encyclopedia of Genes and Genomes(KEGG)途径的宏基因组功能组成。最新开发PICRUSt2软件包可以基于任意的OTU/ASV表直接预测宏基因组功能。R package Tax4Fun可以基于SILVA数据库预测微生物群的KEGG功能能力。原核分类群的功能注释(FAPROTAX)管道基于已发表的代谢和生态功能(如硝酸盐呼吸、铁呼吸、植物病原体和动物寄生虫或共生体)进行功能注释,使其对环境、农业和动物微生物组研究有用。BugBase是一个扩展的绿色基因数据库,用于预测表型,如耐氧性、革兰氏染色和致病潜力;该数据库主要用于医学研究。
Metagenomic analysis
与扩增子相比,鸟枪宏基因组可以直接提供功能基因图谱,并达到更高的分类注释分辨率。然而,由于数据量大,大多数软件仅适用于Linux系统,并且需要大量的计算资源来进行分析。为了便于软件安装和维护,我们建议使用带有BioConda通道的包管理器Conda来部署宏基因组分析管道。由于宏基因组分析是计算密集型的,因此最好并行运行多个任务/样本,这需要GNU parallel等软件来进行队列管理。
Illumina HiSeqX/NovaSeq系统通常产生用于宏基因组测序的PE150读数,而BGI-Seq500产生的读数处于PE100模式。宏基因组分析的第一个关键步骤是质量控制和从原始读数中去除宿主污染,这需要[KneedData管道](https://bitbucket.org/ biobakery/kneaddata)和Bowtie 2的组合。Trimmomatic是Illumina测序数据的灵活质量控制软件包,可用于修剪低质量序列、文库引物和适配器。使用Bowtie 2映射到宿主基因组的读数被视为受污染的读数并过滤掉。KneedData是一个集成的管道,包括Trimmomatic、Bowtie 2和相关脚本,可用于质量控制、删除主机派生的读取和输出干净的读取(图2B)
宏基因组分析的主要步骤是使用基于读数和/或基于组装的方法将干净的数据转换为分类和功能表。基于读取的方法将干净的读取与精心策划的数据库和输出特征表对齐(图2B)。MetaPhlAn2是一种常用的分类分析工具,它将宏基因组读数与预定义的标记基因数据库进行比对,以进行分类。Kraken 2对NCBI非冗余数据库中的序列进行精确的k-mer匹配,并使用最低共同祖先(lowest common ancestor, LCA)算法进行分类。关于20种分类学分类工具的基准测试综述,请参见Ye et al. (2019)。HUMAnN2是一种广泛使用的功能分析软件,也可用于探索样本内和样本间的贡献多样性(物种对特定功能的贡献)。MEGAN是一款跨平台图形用户界面(GUI)软件,用于进行分类和功能分析(表1)。此外,还提供了各种宏基因组基因目录,包括从人类肠道、小鼠肠道、鸡肠道、牛瘤胃。这些定制的数据库可以用于适当研究领域的分类和功能注释,从而实现高效、精确、快速的分析。
基于组装的方法使用MEGAHIT或metaSPAdes等工具将干净的读数组装成重叠群(图2B)。MEGAHIT用于使用很少的计算机内存快速组装大型复杂的宏基因组数据集,而metaSPAdes可以生成更长的重叠群,但需要更多的计算资源。然后使用metaGeneMark或Prokka鉴定组装重叠群中存在的基因。必须使用CD-HIT等工具去除来自单独组装的重叠群的冗余基因。最后,可以使用基于比对的工具(如Bowtie 2)或无比对的方法(如Salmon)生成基因丰度表。数百万个基因通常存在于宏基因组数据集中。这些基因必须组合成功能注释,如KEGG Orthology(KO)、modules和pathways,代表一种降维形式。
此外,宏基因组数据可用于挖掘基因簇或组装微生物基因组草案。抗SMASH数据库用于识别、注释和可视化参与次级代谢产物生物合成的基因簇。Binning是一种可以用于在宏基因组数据中恢复部分或完整细菌基因组的方法。可用的装仓工具包括CONCOCT,MaxBin 2和MetaBAT2。Binning工具根据四核苷酸频率和重叠群丰度将重叠群聚类到不同的Binning(基因组草案)中。进行重新组装以获得更好的bins。我们建议使用binning管道,如MetaWRAP或DAStool,它们集成了几个binning软件包,以获得精细的binning结果和更完整的基因组,污染更少。这些管道还提供了用于评估和可视化的有用脚本。为了对宏基因组实验和分析进行更全面的综述,我们推荐Quince et al. (2017)。