一、基本概念
- 高通量测序技术(High-throughput sequencing)又称“下一代”测序技术("Next-generation" sequencing technology)或者”深度测序“(Deep Sequening),以能一次并行对几十万到几百万条DNA分子进行序列测定和一般读长较短等为标志。
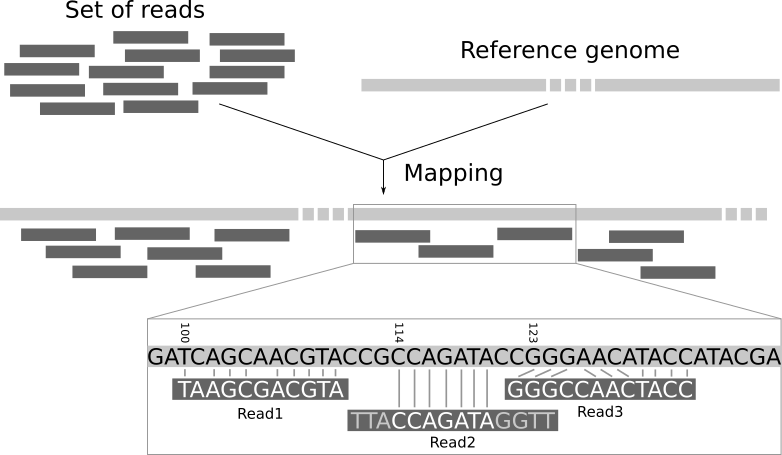
- Reads Mapping:指将测序得到的DNA片段——
reads,定位到参考基因组上。 - Genotyping:是通过使用生物学分析检查个体的DNA序列并将其与另一个个体的序列或参考序列进行比较来确定个体的遗传组成(基因型)差异的过程
- SNP:单核苷酸多态性是单的取代核苷酸发生在一个特定位置的基因组,其中每个变异存在于水平人与人之间的比例为0.5%
二、Reads Mapping
reads mapping克服了深度测序产生Reads过短的技术困难。
reads mapping本质上来说,是序列比对问题。相比Needleman-Wunsch及Smith-Waterman的不同之处在于:
- 长度相差较大,Reads的长度通常不超过100bp,而参考基因组却通常在上百Mb。
- 对于Reads相当于是全局比对
- 对于参考基因组相当于是局部比对
此时可以使用工具BWA这样的工具来将Reads定位到参考基因组,接下来就是安装bwa:
- 下载bwa官网,比较开心的,
bwa是开源的 - 编译、安装,只需在terminal输入
make,就可以生成可执行的bwa程序
算法的更多细节见:北京大学生物信息学课程
三、遗传变异的鉴定
Genotype and SNP calling from next-generation sequencing data
当我们将测序得到的reads定位到参考基因组之后,就可以开始鉴定遗传变异
根据遗传变异的尺寸,将之分为:
- 单核苷酸变异SNV(single nucleotide various)
- SNP 碱基替换(生物分析常用)
- indel
- 涉及多个核苷酸的结构变异SV(structure variation)
- 插入、删除、倒转、异位、拷贝数目变异
genotyping可以使用软件工具GATK