
图片alt
Reconstructing the full-length transcriptome from short reads enabling the discovery of novel transcripts and splicing isoforms
https://www.youtube.com/watch?v=D3PSaxhOVlU
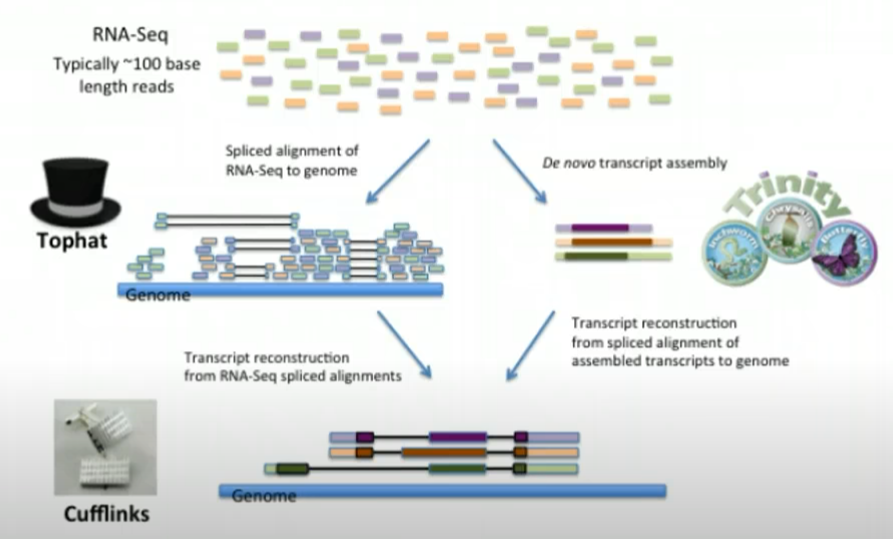
图片alt
转录组测序数据组装
获得高质量的测序数据之后,需要对其进行序列组装。Trinity是一款专门为高通量转录组测序设计的组装软件。转录本测序深度除了受测序数据量等影响,还与该转录本的表达丰度有关。测序深度会直接影响组装的好坏。为了使各样品中表达丰度较低的转录本组装得更完整,对于同物种的测序样品推荐合并组装可以间接增加测序深度,从而使转录结果更完整,同时也有利于后续的数据分析;而对于不同物种的样品,由于基因组间存在差异,推荐采用分别组装或分开分析。
转录组测序文库质量评估
Unigene功能注释
使用BLAST软件将Unigene序列与NR、Swiss-Prot、GO、COG、KOG、eggNOG4.5、KEGG数据库比对,使用KOBAS2.0得到Unigene在KEGG中的KEGG Orthology结果,预测完Unigene的氨基酸序列之后使用HMMER软件与Pfam数据库比对,获得Unigene的注释信息。
基因结构分析
编码区序列预测
TransDecoder软件基于开放阅读框(Open Reading Frame,ORF)长度、对数似然函数值(Log-likelihood Score)、氨基酸序列与Pfam数据库蛋白质结构域序列的比对等信息,能够从转录本序列中识别可靠的潜在编码区序列(Coding Sequence,CDS),是Trinity和Cuffinks等软件官方推荐的CDS预测软件。
简单重复序列分析
MISA(MIcroSAtellite identification tool)是一款鉴定简单重复序列(Simple Sequence Repeat,SSR)的软件,其参考网址见附表。它可以通过对Unigene序列的分析,鉴定出6种类型的SSR:单碱基(Mono-nucleotide)重复SSR、双碱基(Di-nucleotide)重复SSR、三碱基(Tri-nucleotide)重复SSR、四碱基(Tetra-nucleotide)重复SSR、五碱基(Penta-nucleotide)重复SSR和六碱基(Hexa-nucleotide)重复SSR。
SNP分析
利用针对RNA-Seq的比对软件STAR对每个样本的Reads与Unigene序列进行比对,并通过GATK针对RNA-Seq的SNP识别(SNP Calling)流程,识别单核苷酸多态性(Single Nucleotide Polymorphism,SNP)位点。进而可以分析这些SNP位点是否影响了基因的表达水平或者蛋白产物的种类。