
图片alt
Mapped within the insert size and in correct orientation:
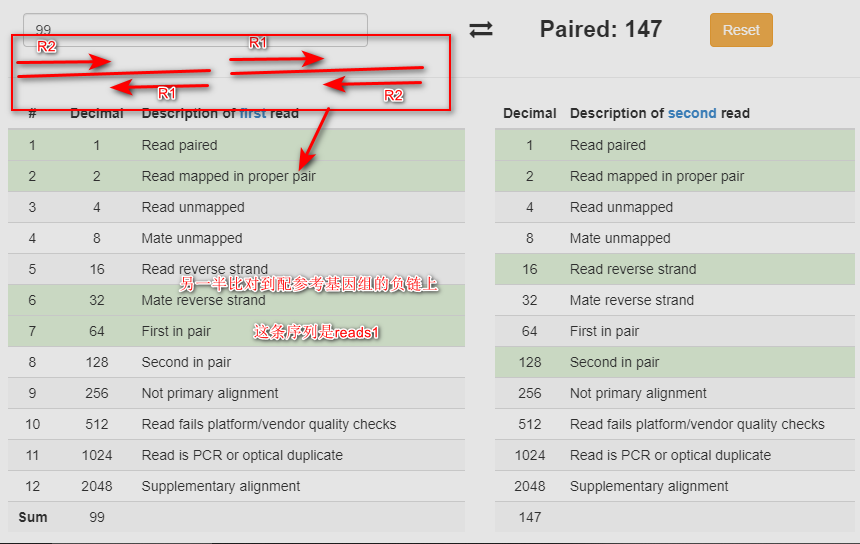
图片alt
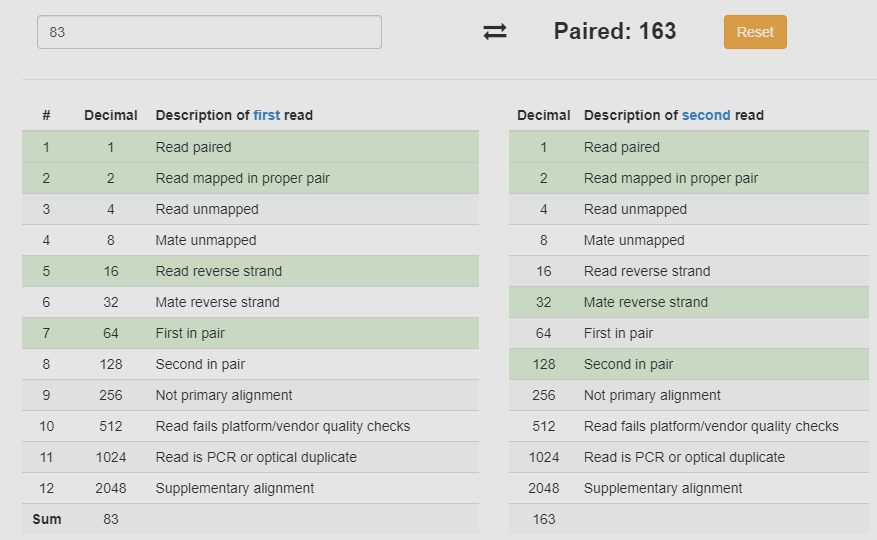
图片alt
Tag
- NM:i:0: number of reads
- MD:Z:60A: 60 match, A mutation
随机读取基因组的任意位置
samtools index -@ 2 input.bam
samtools view -h input.bam char:5000-5500
deuplication
DNA remove duplications by Picard
RNA remove duplications by UMI
去重的经验
- 比较好的解决方案是文库构建时加入UMI或barcode
- RNA表达量分析,一般不去重
- DNA数据分析一般去重
- Chip-seq
- m6A-seq
- ATAC-seq, DNase-seq, MNase-seq
- Hi-C
- BS-seq
reference
https://samformat.info/sam-format-flag