热图的标准化
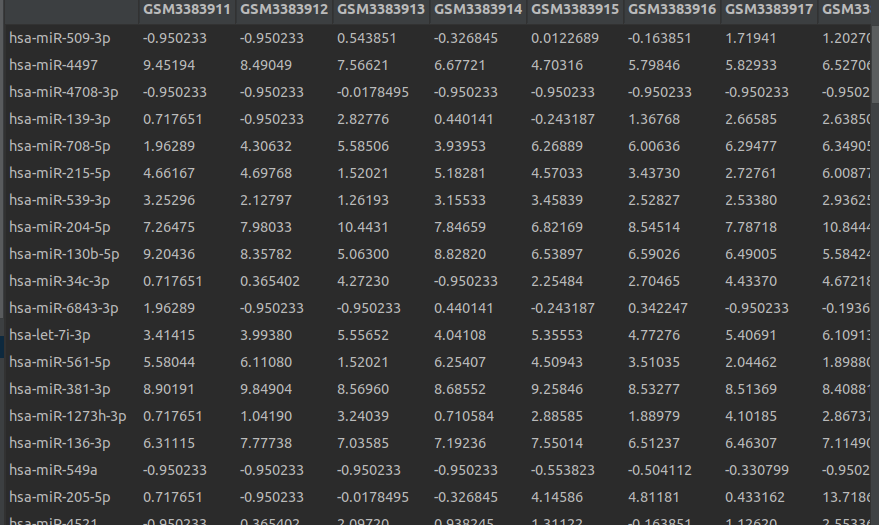
未表准化之前的数据
图片alt
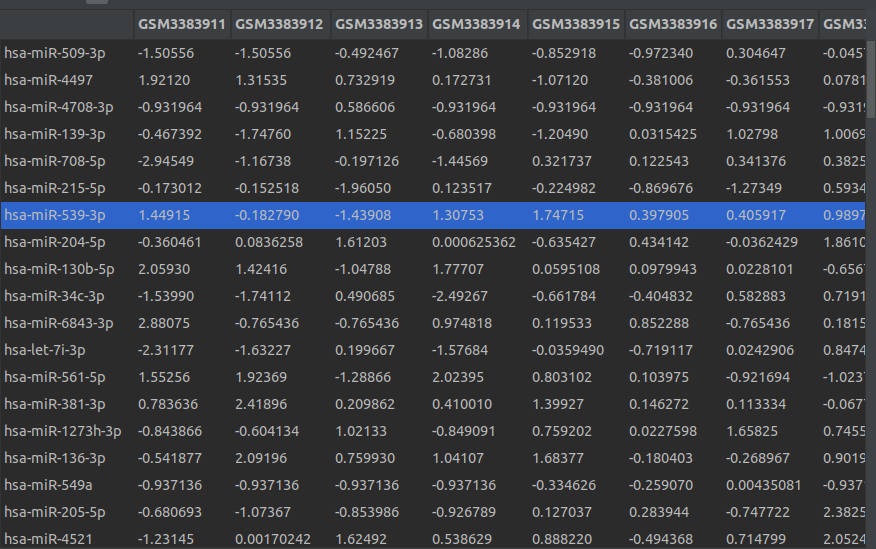
使用pheatmap(scale="row")标准化的数据
scale <- "row"
mat = switch(scale, none = mat, row = scale_rows(mat), column = t(scale_rows(t(mat))))
function(x) {
m = apply(x, 1, mean, na.rm = T)
s = apply(x, 1, sd, na.rm = T)
return((x - m) / s)
}
图片alt
标准化的意义:一个基因减去其在所有样品表达的均值然后除以其在所有样品表达值的标准差,改变每个基因的表达,使该基因在所有细胞中的平均表达为0,缩放每个基因的表达,使样品间的方差为1,使各个基因在下游分析中给予同等权重,因此高表达基因不会占主导地位,使得基因之间可以互相比较
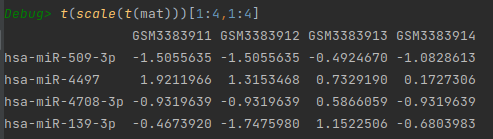
也可以使用scale进行标准化,但是scale默认是对列进行表转化,因此需要转置
图片alt
单细胞的标准化
Normalizing the data
从数据集中删除不需要的cells后,下一步是normalize the data。默认情况下,使用global-scaling normalization method (LogNormalize),该方法将每个cell expression测量值标准化为total expression,将其乘以scale factor(默认情况下为10000),然后对结果进行log-transforms。标准化值存储在pbmc[["RNA"]]@data

pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
NormalizeData <- function(object, ...) {
UseMethod(generic = "NormalizeData", object = object)
}
class(object) # class is Assay
当object的class是Assay时,调用NormalizeData.Assay,而NormalizeData.Assay 调用NormalizeData.default(new.data的值)
NormalizeData.Assay <- function(){
object <- SetAssayData(object = object, slot = "data", new.data = NormalizeData(object = GetAssayData(object = object, slot = "counts"), normalization.method = normalization.method, scale.factor = scale.factor, verbose = verbose, margin = margin, ...))
return(object)
}
当object的class是dgCMatrix时,调用NormalizeData.default,而NormalizeData.default主要是调用LogNormalize
NormalizeData(object = GetAssayData(object = pbmc, slot = "counts"))
GetAssayData(object = pbmc, slot = "counts") # class is dgCMatrix
NormalizeData.default <- function(object, normalization.method = "LogNormalize", scale.factor = 10000, margin = 1, block.size = NULL, verbose = TRUE, ...){
CheckDots(...)
if (is.null(x = normalization.method)) {
return(object)
}
normalized.data <- if (nbrOfWorkers() > 1) {
......
} else {
switch(EXPR = normalization.method, # LogNormalize
LogNormalize = LogNormalize(data = object,
scale.factor = scale.factor,
verbose = verbose),
CLR = CustomNormalize(data = object,
custom_function = function(x) {
return(log1p(x = x / (exp(x = sum(log1p(x = x[x > 0]), na.rm = TRUE) / length(x = x)))))
},
margin = margin,
verbose = verbose),
RC = RelativeCounts(data = object, scale.factor = scale.factor, verbose = verbose),
stop("Unknown normalization method: ", normalization.method))
}
return(normalized.data)
}
LogNormalize主要是调用LogNorm
LogNormalize <- function(data, scale.factor = 10000, verbose = TRUE) {
if (is.data.frame(x = data)) {
data <- as.matrix(x = data)
}
if (!inherits(x = data, what = "dgCMatrix")) {
data <- as(object = data, Class = "dgCMatrix")
}
if (verbose) {
cat("Performing log-normalization\n", file = stderr())
}
norm.data <- LogNorm(data, scale_factor = scale.factor, display_progress = verbose)
colnames(x = norm.data) <- colnames(x = data)
rownames(x = norm.data) <- rownames(x = data)
return(norm.data)
}
这里使用了Rcpp的Eigen库,一个Rcpp调用C++ Eigen库的demo
R/RcppExports.R:21
LogNorm <- function(data, scale_factor, display_progress = TRUE) {
.Call('_Seurat_LogNorm', PACKAGE = 'Seurat', data, scale_factor, display_progress)
}
src/RcppExports.cpp:77
// LogNorm
Eigen::SparseMatrix<double> LogNorm(Eigen::SparseMatrix<double> data, int scale_factor, bool display_progress);
RcppExport SEXP _Seurat_LogNorm(SEXP dataSEXP, SEXP scale_factorSEXP, SEXP display_progressSEXP) {
BEGIN_RCPP
Rcpp::RObject rcpp_result_gen;
Rcpp::traits::input_parameter< Eigen::SparseMatrix<double> >::type data(dataSEXP);
Rcpp::traits::input_parameter< int >::type scale_factor(scale_factorSEXP);
Rcpp::traits::input_parameter< bool >::type display_progress(display_progressSEXP);
rcpp_result_gen = Rcpp::wrap(LogNorm(data, scale_factor, display_progress));
return rcpp_result_gen;
END_RCPP
}
Scaling the data
总结
用机器学习中的术语来说,每一个基因相当于特征,例如影响银行贷款的年龄、工资等自变量,银行贷款的因变量是分类变量的话,就是能或不能贷款给你,而基因的因变量就是表型(肿瘤的发生,转移)。