
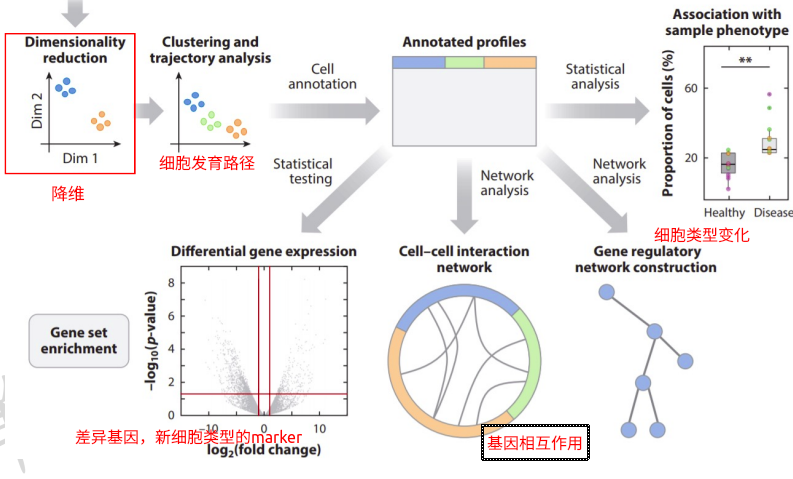
图片alt
图片alt
先决条件
- 软件
- 比对软件:STAR
- 提取UMI和cell barcode:umi_tools
- 质控fasta文件:cutadapt
- 基因计数:featureCounts (安装subread)
- seurat
- Current best practices in single-cell RNA-seq analysis: a tutorial
- 参考基因组

从fasta文件到count matrix
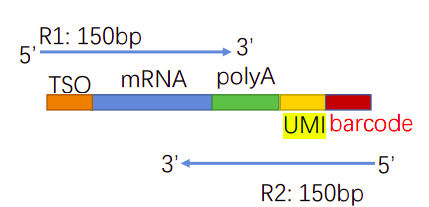
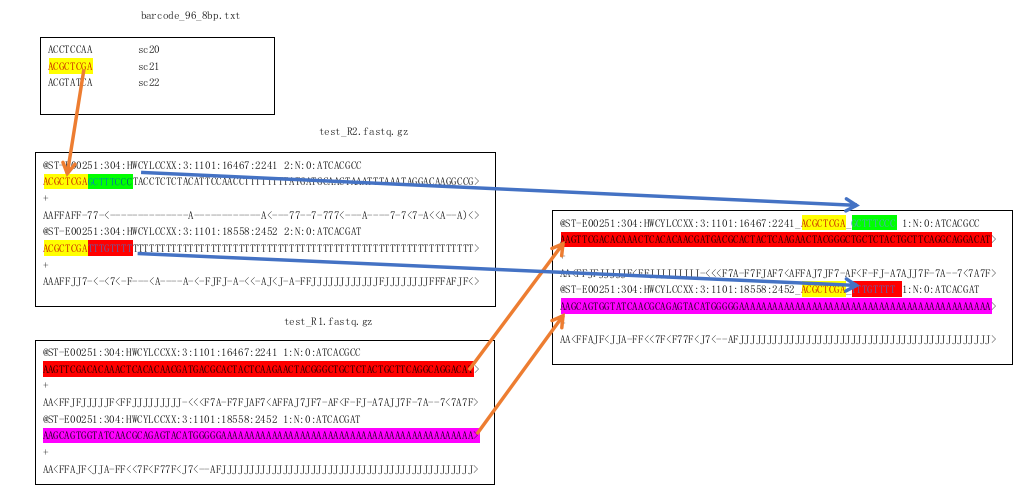
提取 cell barcode和unique molecular identifier
从一个fasta文件中读取中提取 cell barcode和UMI ,并将其添加到相应的fasta文件的相同reads的header中
图片alt
umi_tools extract
# C = cell barcode position; N = UMI position
umi_tools extract --bc-pattern=CCCCCCCCNNNNNNNN \ # barcode+umi
--stdin test_R2.fastq.gz \ #
--stdout test.R1.extracted.fq.gz \
--read2-stdout \
--read2-in test_R1.fastq.gz \
--whitelist=barcode_96_8bp.txt # barcode细胞类型文件
图片alt
数据质控
cutadapt \
-g TGGTATCAACGCAGAGTACATGGG \ # 去除TSO GGG
-a AAAAAAAAAAAAAAA \ # 去除polyA
test.R1.extracted.fq.gz \
-m 37 \ # 最短reads要求37bp
-o test.R1.clean.fq.gz
Trim Galore:质控,去接头,删除 RRBS 序列文件的偏向甲基化位置
数据比对
STAR
构建index
STAR --runThreadN 20 --runMode genomeGenerate --genomeDir ./star --genomeFastaFiles fasta/genome.fa --sjdbGTFfile genes/genes.gtf
比对
STAR --runThreadN 4 \
--genomeDir $genomeDir \ # 参考基因组
--readFilesIn test.R1.clean.fq.gz \
--readFilesCommand zcat \
--outFilterMultimapNmax 1 \ #比对时允许的最大错配数
--outFileNamePrefix test. \
--outSAMtype BAM SortedByCoordinate
质控:test.Log.final.out, bam文件: test.Aligned.sortedByCoord.out.bam
转录本定量
featureCounts -a $gtf \
-o gene_assigned \
-R BAM test.Aligned.sortedByCoord.out.bam \
-T 4
test.Aligned.sortedByCoord.out.bam.featureCounts.bam,加上一列tag标记基因名字 XT:Z:ENSG00000206172
整理bam文件
samtools sort -m 15000000000 test.Aligned.sortedByCoord.out.bam.featureCounts.bam -o test.assigned_sorted.bam;
samtools index test.assigned_sorted.bam
获取单细胞基因表达矩阵
umi tools count
适用于大多数单细胞 RNA-Seq 方法,该工具仅设计用于在扩增后发生片段化的文库制备方法。 由于精确的映射坐标不再为此类文库制备提供信息,因此将其简化为基因为单位统计测到的UMI数。
umi_tools count --per-gene --gene-tag=XT \
--per-cell --wide-format-cell-counts \
-I test.assigned_sorted.bam -S test.UMI_counts.tsv
从表达矩阵开始(鉴定细胞类型)

图片alt
质量控制(过滤低质量细胞,过滤不表达基因)
- 过滤低质量细胞
- 基因数:>200,1k, 2k
- 线粒体比例:
<5%- mtDNA%: the fraction of mitochondrial counts per cell High numbers of mitochondrial transcripts are indicators of cell stress, and therefore mtDNA% is a measurement associated with apoptotic, stressed and low-quality cells. (Daniel Osorio and James J. Cai, Bioinformatics, 2020)
- count数:doublets
- 过滤不表达基因
- 细胞数:
>3
- 细胞数:
降维聚类后低质量细胞会聚在一起,观察这群细胞的基因数,UMI数和线粒体比例,取合理范围进行质控
数据校正、标准化normalization
使得细胞之间可以相互比较(细胞的测序深度相同)
- 测序深度(sequencing depth)标准化
- Downsampling:抽取等量reads
- CPM (count per million)
- CPM=106*Cij/∑iCij(每个细胞测了10000个reads)
- data=log((CPM/10) + 1)
- 批次效应(batch effect)
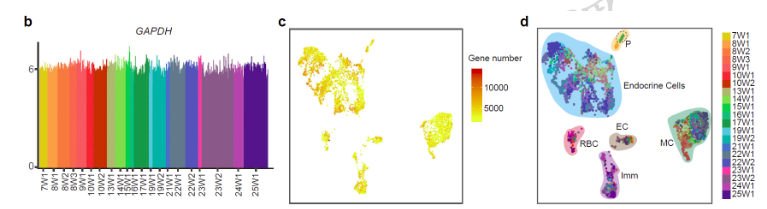
证明数据质量好
- 基因数和UMI数比较大
- 没有明显的批次效应,细胞分群不是来自不同样本,cell type的聚类是正确的
- 管家基因的表达一致
- 同一个细胞类型里有来源不同的样品
- 测序深度或基因数没有明显影响细胞聚类
图片alt
选取高异质性基因 highly variable genes
- 细胞间高异质性基因:它们在某些细胞中高度表达,而在其他细胞中低表达
- 大部分的基因表达波动是由于技术误差造成的。波动特别大的通常和细胞生物学功能有关
- 绘制基因平均表达量(log转换)和方差的点图,拟合一条曲线,近似表示该表达量的基因受技术误差导致的方差。远高于拟合曲线的点表示有意义的生物水平造成的基因表达方差。
归一化
- 基因归一化是指一个基因减去其在所有样品表达的均值然后除以其在所有样品表达值的标准差
- 改变每个基因的表达,使该基因在所有细胞中的平均表达为0
- 缩放每个基因的表达,使细胞间的方差为1
- 使各个基因在下游分析中给予同等权重,因此高表达基因不会占主导地位
- 使得基因之间可以互相比较
降维-二维平面可视化 dimension reduction
- 主成分分析法(principal component analysis,PCA)
- t分布随机临近嵌入(t-distributed stochastic neighbor embedding, t-SNE):体现数据局部结构特征
- 均匀流形近似与投影(uniform manifold approximation and projection, UMAP):UMAP可以更好的保留样本的邻近信息和聚类精度,体现数据全局结构特征,有利于发现聚类的生物和临床特征。强烈推荐!
- Dimensionality reduction by UMAP to visualize physical and genetic interactions
分群 clustering
- FindNeighbors() 将先前定义的数据集维度(前15 个PC)作为输入,基于PCA 空间中的欧几里德距离构建一个KNN (K-近邻算法)图,并基于其局部邻域中的共享重叠细化任意两个单元之间的边权重
- FindClusters()聚类细胞,应用模块化优化技术,例如Louvain算法(默认),将细胞迭代分组在一起。调整分辨率参数resolution来控制分群个数,resolution越大分群越多,较大的数据集通常需要增加resolution(>2)

图片alt
标志基因定义细胞类型:biomarker
- 转录因子:影响谱系lineage分化,遗传操作可控制该细胞类型
- 表面蛋白:抗体富集该种细胞类型
- 特异表达基因或基因集组合
- 如何找标志基因
- 看文献收集:review文章;相关领域单细胞文章。
- 数据库
- 实验室传承
- 红细胞:c('HBG1','HBG2','HBQ1','HBA1','HBA2','HBE1','HBD','HBM','HBZ','HBB')
- 性别:c('XIST','UTY','DDX3Y','TTTY15','EIF1AY','TXLNGY','RPS4Y1','KDM5D','USP9Y’)
- 上皮细胞:c("EPCAM","CDH1","KRT5","KRT7","KRT8","KRT18","KRT19","KRT14")
- 免疫细胞: c("PTPRC"); T cell; NK cell; B cell; Macrophage; Mast cell; Dendritic cell etc.al
- 成纤维细胞: c("THY1", "DCN" ,"PODN", "COL1A1","COL1A2", "COL3A1","COL6A1")
- 内皮细胞:c("PECAM1","VWF", "ENG","CDH5")
将已知的标志基因map到分群上

图片alt
CellMarker
celltaxonomy
寻找差异基因 differentially expressed genes
- 已知的marker gene一般都是显著的差异基因,进一步证明分群正确
- 寻找新的标志基因
- p_val :Wilcox
- avg_log2FC: >=1.5或2,取决于差异基因数
- pct.1:在本群表达细胞的比例,越高越好
- pct.2:在非本群表达细胞的比例,越低越好
- p_val_adj:<=0.05; bonferronicorrection based on the total number of genes in the dataset
功能注释
- R package: clusterprofiler
- metascape
f
图片alt

图片alt