- Step-by-step network construction and module detection
- Relating modules to external clinical traits
- One-step network construction and module detection
- 参考
Step-by-step network construction and module detection
dim:134*3600
MMT00000044 MMT00000046 MMT00000051
F2_2 -1.81e-02 -0.0773 -0.0226
F2_3 6.42e-02 -0.0297 0.0617
F2_14 6.44e-05 0.1120 -0.1290
1. Co-expression similarity and adjacency
现在,我们使用soft thresholding 6 计算adjacencies:
softPower = 6;
adjacency = adjacency(datExpr, power = softPower);
dim:3600*3600
MMT00000044 MMT00000046 MMT00000051
MMT00000044 1.000000e+00 2.997850e-09 6.581419e-07
MMT00000046 2.997850e-09 1.000000e+00 3.216588e-02
MMT00000051 6.581419e-07 3.216588e-02 1.000000e+00
2. Topological Overlap Matrix (TOM)
为了最小化noise和虚假关联(spurious associations)的影响,我们将邻接(adjacency)转换为拓扑重叠矩阵(Topological Overlap Matrix),并计算相应的相异度(dissimilarity):
TOM = TOMsimilarity(adjacency);
dissTOM = 1-TOM
dim:3600*3600
[,1] [,2] [,3]
[1,] 1.0000000000 0.0007954678 0.0006276637
[2,] 0.0007954678 1.0000000000 0.0478588481
[3,] 0.0006276637 0.0478588481 1.0000000000
3. Clustering using TOM
我们现在使用hierarchical clustering 来生成基因的hierarchical clustering tree(树状图)。 请注意,我们使用的函数hclust提供了比标准hclust函数更快的分层聚类例程。
geneTree = fastcluster::hclust(as.dist(dissTOM), method = "average");
plot(geneTree,
xlab="",
sub="",
main = "Gene clustering on TOM-based dissimilarity",
labels = FALSE, hang = 0.04);

图片alt
每片叶子,即一条短的垂直线,对应一个基
minModuleSize = 30;
dynamicMods = cutreeDynamic(dendro = geneTree,
distM = dissTOM,
deepSplit = 2,
pamRespectsDendro = FALSE,
minClusterSize = minModuleSize);
table(dynamicMods)
dynamicMods
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
88 614 316 311 257 235 225 212 158 153 121 106 102 100 94 91 78 76 65 58 58 48 34
该函数返回22个模块,标记为1–22最大到最小。标签0保留给未分配的基因。上面的命令列出了模块的大小。
dynamicColors = labels2colors(dynamicMods)
table(dynamicColors)
plotDendroAndColors(geneTree,
dynamicColors,
"Dynamic Tree Cut",
dendroLabels = FALSE,
hang = 0.03,
addGuide = TRUE,
guideHang = 0.05,
main = "Gene dendrogram and module colors")
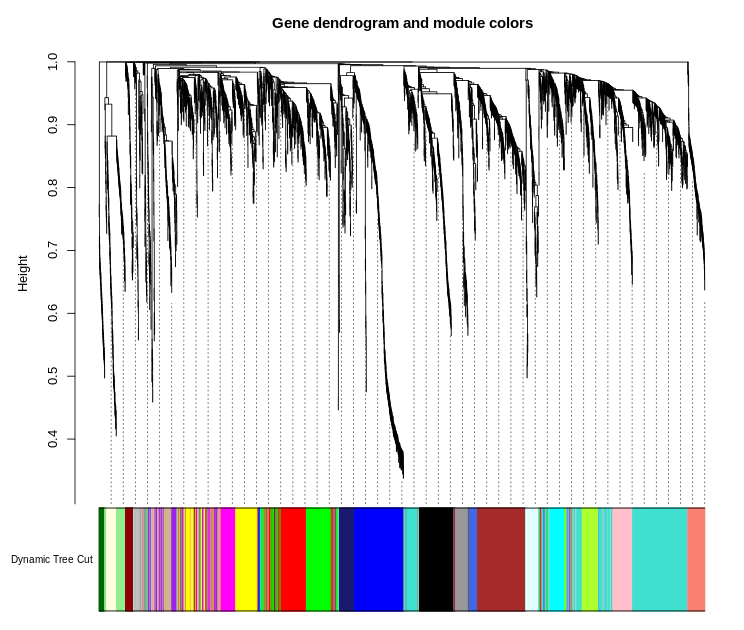
图片alt
4. Merging of modules whose expression profiles are very similar
动态树切割可以识别expression profiles非常相似的模块。合并这些模块可能是谨慎的,因为它们的基因是高度共表达的。为了量化整个模块的共表达相似性,我们计算它们的特征基因,并根据它们的相关性对它们进行聚类:
MEList = moduleEigengenes(datExpr, colors = dynamicColors) # Calculate eigengenes
MEs = MEList$eigengenes
MEs[1:3,1:3]

图片alt
> MEs[1:3,1:3]
MEblack MEblue MEbrown
F2_2 0.01390248 0.0410177922 0.007072125
F2_3 0.06667534 -0.0009540238 0.072447744
F2_14 0.06671191 -0.0841292811 0.062700422
MEDiss = 1-cor(MEs);
MEDiss[1:3,1:3]
> MEDiss[1:3,1:3]
MEblack MEblue MEbrown
MEblack 0.0000000 1.524265 0.4051713
MEblue 1.5242653 0.000000 1.1824857
MEbrown 0.4051713 1.182486 0.0000000
METree = hclust(as.dist(MEDiss), method = "average");
plot(METree,
main = "Clustering of module eigengenes",
xlab = "",
sub = "")
# 我们选择0.25的高度切割,对应于0.75的相关性,以进行合并:
MEDissThres = 0.25 #0.25
abline(h=MEDissThres, col = "red")
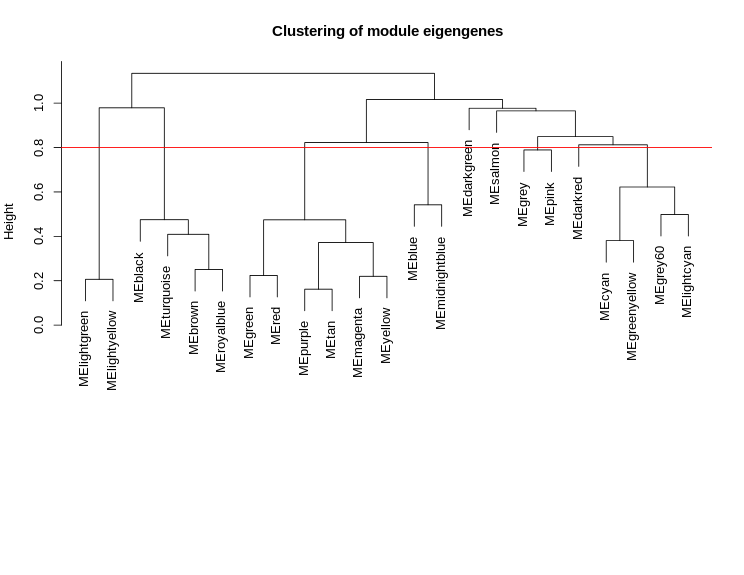
图片alt
merge = mergeCloseModules(datExpr, dynamicColors, cutHeight = MEDissThres, verbose = 3)

图片alt
plotDendroAndColors(geneTree,
cbind(dynamicColors, mergedColors),
c("Dynamic Tree Cut", "Merged dynamic"),
dendroLabels = FALSE,
hang = 0.03,
addGuide = TRUE,
guideHang = 0.05)

图片alt
Relating modules to external clinical traits
Quantifying module–trait associations
mergedMEs = merge$newMEs;
MEs = orderMEs(mergedMEs)
MEs[1:3,1:3]
> MEs[1:3,1:3]
MElightgreen MEblack MEturquoise
F2_2 -0.022004340 0.01390248 0.02035156
F2_3 -0.004099771 0.06667534 0.03761149
F2_14 0.593793143 0.06671191 0.19275250
moduleTraitCor = cor(MEs, datTraits, use = "p");
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nrow(datExpr));
> moduleTraitCor[1:3,1:3]
weight_g length_cm ab_fat
MElightgreen -0.01741811 0.08035027 -0.005618515
MEblack -0.31277458 -0.15465924 -0.273439183
MEturquoise -0.27364499 -0.14615075 -0.328091062
> moduleTraitPvalue[1:3,1:3]
weight_g length_cm ab_fat
MElightgreen 0.8416719204 0.35605884 0.9486276885
MEblack 0.0002337232 0.07437667 0.0013896872
MEturquoise 0.0013776808 0.09198325 0.0001088202
textMatrix = paste(signif(moduleTraitCor, 2), "\n(", signif(moduleTraitPvalue, 1), ")", sep = "");
dim(textMatrix) = dim(moduleTraitCor)
> textMatrix[1:3,1:3]
[,1] [,2] [,3]
[1,] "-0.017\n(0.8)" "0.08\n(0.4)" "-0.0056\n(0.9)"
[2,] "-0.31\n(2e-04)" "-0.15\n(0.07)" "-0.27\n(0.001)"
[3,] "-0.27\n(0.001)" "-0.15\n(0.09)" "-0.33\n(1e-04)"
par(mar = c(6, 8.5, 3, 3));
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
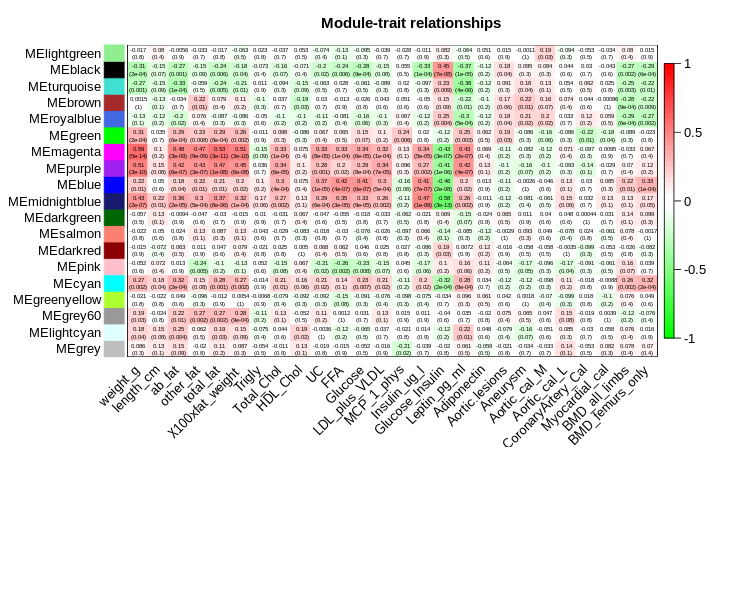
图片alt
Gene Significance and Module Membership
我们通过将 Gene Significance GS定义为基因与性状之间的相关性(绝对值),来量化单个基因与我们感兴趣的性状(体重)之间的关联。对于每个模块,我们还定义了 module membership MM的定量度量,即module eigengene和gene expression profile的相关性
geneModuleMembership = as.data.frame(cor(datExpr, MEs, use = "p"));
MMPvalue = as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nrow(datExpr)));
modNames = substring(names(MEs), 3)
names(geneModuleMembership) = paste("MM", modNames, sep="");
names(MMPvalue) = paste("p.MM", modNames, sep="");
> geneModuleMembership[1:3,1:3]
MMlightgreen MMblack MMturquoise
MMT00000044 0.02485814 0.06893329 -0.1600427
MMT00000046 0.05952419 0.17672545 0.7885263
MMT00000051 -0.07295982 -0.32001047 -0.7469499
> MMPvalue[1:3,1:3]
p.MMlightgreen p.MMblack p.MMturquoise
MMT00000044 0.7755660 0.4286949897 6.471859e-02
MMT00000046 0.4944849 0.0410835122 1.184869e-29
MMT00000051 0.4021566 0.0001637068 3.690993e-25
计算基因表达量和weight之间的相关性
weight = as.data.frame(datTraits$weight_g)
names(weight) = "weight"
geneTraitSignificance = as.data.frame(cor(datExpr, weight, use = "p"));
GSPvalue = as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nrow(datExpr)));
names(geneTraitSignificance) = paste("GS.", names(weight), sep="");
names(GSPvalue) = paste("p.GS.", names(weight), sep="");
> geneTraitSignificance
weight
MMT00000044 -6.937286e-02
MMT00000046 -7.525085e-02
MMT00000051 2.434278e-01
MMT00000076 1.712195e-01
我们在green模块中绘制了 Gene Significance与Module Membership关系的散点图
module = "green"
column = match(module, modNames);
moduleGenes = mergedColors==module;
MM <- abs(geneModuleMembership[moduleGenes, column])
GS <- abs(geneTraitSignificance[moduleGenes, 1])
verboseScatterplot(MM,
GS,
xlab = paste("Module Membership in", module, "module"),
ylab = "Gene significance for body weight",
main = paste("Module membership vs. gene significance\n"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)

图片alt
geneInfo0 = data.frame(symbol = names(datExpr),
moduleColor = mergedColors,
geneTraitSignificance,
GSPvalue)
modOrder = order(-abs(cor(MEs, weight, use = "p")));
for (mod in 1:ncol(geneModuleMembership))
{
oldNames = names(geneInfo0)
geneInfo0 = data.frame(geneInfo0, geneModuleMembership[, modOrder[mod]],
MMPvalue[, modOrder[mod]]);
names(geneInfo0) = c(oldNames, paste("MM.", modNames[modOrder[mod]], sep=""),
paste("p.MM.", modNames[modOrder[mod]], sep=""))
}
geneOrder = order(geneInfo0$moduleColor, -abs(geneInfo0$GS.weight));
geneInfo = geneInfo0[geneOrder, ]
geneInfo[1:3,1:10]
> geneInfo[1:3,1:10]
symbol moduleColor GS.weight p.GS.weight MM.magenta p.MM.magenta MM.purple p.MM.purple
MMT00030014 MMT00030014 black -0.5797950 2.125494e-13 -0.5549815 3.438475e-12 -0.5512728 5.111937e-12
MMT00035294 MMT00035294 black 0.4608253 2.106843e-08 0.5054773 4.687405e-10 0.5193470 1.280101e-10
MMT00074983 MMT00074983 black 0.4432473 8.165953e-08 0.5947134 3.549372e-14 0.6373045 1.242206e-16
One-step network construction and module detection
net = blockwiseModules(datExpr,
power = 6,
TOMType = "unsigned",
minModuleSize = 30,
reassignThreshold = 0,
mergeCutHeight = 0.25,
numericLabels = TRUE,
pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "femaleMouseTOM",
verbose = 3)
mergedColors = labels2colors(net$colors)
unmergedColors = labels2colors(net$unmergedColors)
# Plot the dendrogram and the module colors underneath
plotDendroAndColors(net$dendrograms[[1]],
cbind(mergedColors[net$blockGenes[[1]]],unmergedColors[net$blockGenes[[1]]]),
c("mergedColors", "unmergedColors"),
dendroLabels = FALSE,
hang = 0.03,
addGuide = TRUE,
guideHang = 0.05)
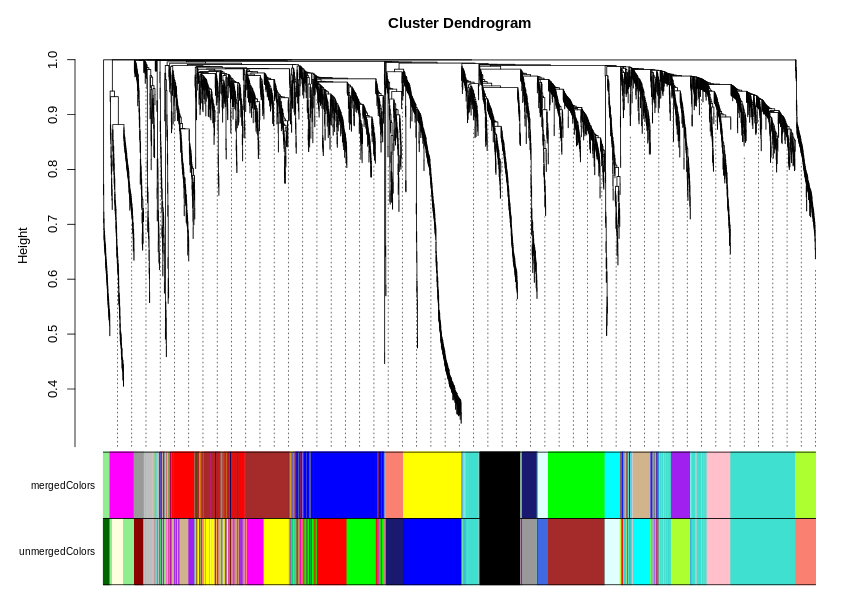
图片alt
library(WGCNA)
rt=read.table("merge.txt",sep="\t",row.names=1,header=T,check.names=F,quote="!")
datExpr = t(rt)
######select beta value######
powers1=c(seq(1,10,by=1),seq(12,30,by=2))
RpowerTable=pickSoftThreshold(datExpr, powerVector=powers1)[[2]]
cex1=0.7
#pdf(file="softThresholding.pdf")
par(mfrow=c(1,2))
plot(RpowerTable[,1], -sign(RpowerTable[,3])*RpowerTable[,2],xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n")
text(RpowerTable[,1], -sign(RpowerTable[,3])*RpowerTable[,2], labels=powers1,cex=cex1,col="red")
# this line corresponds to using an R^2 cut-off of h
abline(h=0.85,col="red")
plot(RpowerTable[,1], RpowerTable[,5],xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n")
text(RpowerTable[,1], RpowerTable[,5], labels=powers1, cex=cex1,col="red")
beta1=9
ADJ= adjacency(datExpr,power=beta1)
vis=exportNetworkToCytoscape(ADJ,edgeFile="edge.txt",nodeFile="node.txt",threshold = 0.8)