1 双端测序reads的方向与转录本的方向问题

图片alt
传统的illumina测序,无法得知哪一个reads的方向与转录本方向一致,哪一个与转录本反向互补
2 junction reads需要正确断开
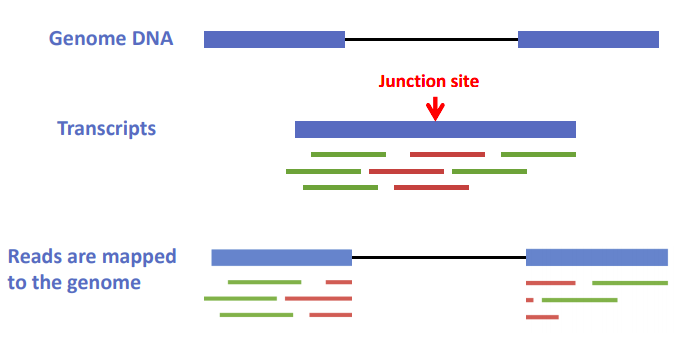
图片alt
具体的解决方法包块:join exon、split reads
2.1 join exon

图片alt
- 构建所有可能的junction的库,即所有可能的外显子拼接
- 可以发现以前未知的exon
- 缺点:不能发现新的exon
2.2 split reads
- 将 junction reads切割为更小的片段mapping到基因组
- 可以发现新的exon
3 TopHat
tophat [options]* <genome_index_base> <reads1_1[,...,readsN_1]> [reads1_2,...readsN_2]
-r/--mate-inner-dist <int>:插入片段长度,将RNA打断为300bp的小片段,两端测序75bp的reads,中间插入片段长度为150bp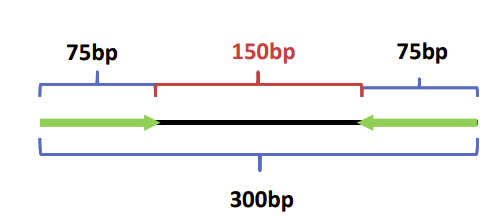
图片alt
--mate-std-dev <int>:插入片段长度的标准差-G/--GTF <GTF/GFF3 file>:基因组已有的注释文件,首先将reads回帖到转录组,没有回帖到转录组的回帖到基因组,junction reads可以直接回帖到转录本--library-type:默认是fr-unstranded,确定数据是是否为either fr-firststrand or fr-secondstrand
3.1 先决条件
wget ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE32nnn/GSE32038/suppl/GSE32038%5Fsimulated%5Ffastq%5Ffiles%2Etar%2Egz
wget ftp://ftp.ensembl.org/pub/release 90/fasta/drosophila_melanogaster/dna/Drosophila_melanogaster.BDGP6.dna.toplevel.fa.gz
3.2 建立参考基因组索引
- Index and annotation downloads
- bowtie2-build
bowtie2-build Drosophila_melanogaster.BDGP6.dna.toplevel.fa genome
3.3 reads回帖到参考基因组
tophat -p 16 -G ../data/reference/Saccharomyces_cerevisiae.R64-1-1.48.gtf -o EV_3 ../data/reference/genome ../data/RAN_seq/SRR1916152.fastq