ceRNAs network analysis using GDCRNATools
GDCRNATools: integrative analysis of protein coding genes, long non-coding genes, and microRNAs in GDC
github
library(GDCRNATools)
####### ceRNA network analysis #######
deLNC <- c('ENSG00000260920','ENSG00000242125','ENSG00000261211')
dePC <- c('ENSG00000043355','ENSG00000109586','ENSG00000144355')
genes <- c(deLNC, dePC)
samples <- c('TCGA-2F-A9KO-01', 'TCGA-2F-A9KP-01',
'TCGA-2F-A9KQ-01', 'TCGA-2F-A9KR-01',
'TCGA-2F-A9KT-01', 'TCGA-2F-A9KW-01')
rnaExpr <- data.frame(matrix(c(2.7,7.0,4.9,6.9,4.6,2.5,
0.5,2.5,5.7,6.5,4.9,3.8,
2.1,2.9,5.9,5.7,4.5,3.5,
2.7,5.9,4.5,5.8,5.2,3.0,
2.5,2.2,5.3,4.4,4.4,2.9,
2.4,3.8,6.2,3.8,3.8,4.2),6,6),
stringsAsFactors=FALSE)
rownames(rnaExpr) <- genes
colnames(rnaExpr) <- samples
mirExpr <- data.frame(matrix(c(7.7,7.4,7.9,8.9,8.6,9.5,
5.1,4.4,5.5,8.5,4.4,3.5,
4.9,5.5,6.9,6.1,5.5,4.1,
12.4,13.5,15.1,15.4,13.0,12.8,
2.5,2.2,5.3,4.4,4.4,2.9,
2.4,2.7,6.2,1.5,4.4,4.2),6,6),
stringsAsFactors=FALSE)
colnames(mirExpr) <- samples
rownames(mirExpr) <- c('hsa-miR-340-5p','hsa-miR-181b-5p',
'hsa-miR-181a-5p', 'hsa-miR-181c-5p',
'hsa-miR-199b-5p','hsa-miR-182-5p')
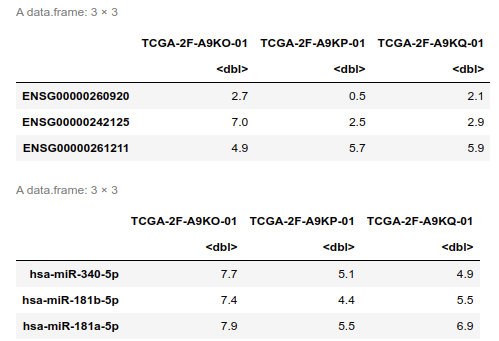
rnaExpr[1:3,1:3]
mirExpr[1:3,1:3]
图片alt
ceOutput <- gdcCEAnalysis(lnc = deLNC,
pc = dePC,
lnc.targets = 'starBase',
pc.targets = 'starBase',
rna.expr = rnaExpr,
mir.expr = mirExpr)
ceOutput

图片alt
# ceOutput2 <- ceOutput[ceOutput$hyperPValue<0.01 &
# ceOutput$corPValue<0.01 & ceOutput$regSim != 0,]
edges <- gdcExportNetwork(ceNetwork = ceOutput, net = 'edges')
nodes <- gdcExportNetwork(ceNetwork = ceOutput, net = 'nodes')
edges
nodes

图片alt
library(ggplot2)
library(ggridges)
head(diamonds)
ggplot(diamonds, aes(x = price, y = cut,fill=cut,
group=cut, height=..density..)) +
geom_density_ridges(scale = 4) +
scale_y_discrete(expand = c(0, 0)) + # will generally have to set the `expand` option
scale_x_continuous(expand = c(0, 0)) + # for both axes to remove unneeded padding
coord_cartesian(clip = "off") + # to avoid clipping of the very top of the top ridgeline
theme_ridges()
Venn
png(
file = "figure/ggvenn.png",
width = 15, height = 15, units = "cm", bg = "white", res = 300
)
ggvenn(
list("TCGA Mutation" = TCGA_mutation, "Sequencing Mutation" = my_mutation),
stroke_size = 0.5, set_name_size = 5, text_size = 5
)
dev.off()
################################################################################
#
# 食管癌GEO ceRNA
#
################################################################################
{
library("optparse")
library(tidyverse)
library(tidyverse)
library(ggrepel)
library(edgeR)
library(pheatmap)
library('SpidermiR')
library("ggvenn")
library(clusterProfiler)
library(org.Hs.eg.db)
library(ggpubr)
library(lattice)
library(survivalROC)
option_list = list(make_option(c("-d","--directory"), type = "character", help = "test"))
args <- parse_args(OptionParser(option_list=option_list))
if(!is.null(args$directory)){
if(!file.exists(args$directory)){
message("请确保 ",args$directory," 存在!")
q()
}
setwd(args$directory)
message("切换工作目录到:",args$directory)
}
if(!file.exists("figures")){
dir.create(file.path("figures"))
}
if(!file.exists("figures/GEO_Volcano_heatmap")){
dir.create(file.path("figures/GEO_Volcano_heatmap"))
}
if(!file.exists("figures/venn")){
dir.create(file.path("figures/venn"))
}
if(!file.exists("figures/survival")){
dir.create(file.path("figures/survival"))
}
if(!file.exists("figures/cox")){
dir.create(file.path("figures/cox"))
}
if(!file.exists("result/enrichment")){
dir.create(file.path("result/enrichment"))
}
}
{
IS_DEBUG=T
LOG_FILENAME= "report_download.txt"
IS_EMPTY=T
LOG_FILENAME= source("/home/wangyang/workspace/bioinfo_analysis/Rscript/tools/tools.R")
}
####################################################
## 公共函数
###################################################
{
getDeg <- function(deg){
res <- deg %>% rownames_to_column("symbol") %>%
dplyr::select(symbol,logFC=logFC,Pvalue=adj.P.Val)
return(res)
}
Pheatmap <- function(deg,expr,filename){
deg_gene <- getDeg(deg)%>%
filter(abs(logFC)>2,Pvalue<0.01)%>%
arrange(desc(abs(logFC)))%>%
pull("symbol")
deg_expr <- expr[deg_gene,]%>%
{log10(cpm(.)+0.01)}
#deg_expr <- t(scale(t(deg_expr)))
#deg_expr[deg_expr > 2] = 2
#deg_expr[deg_expr < -2] = -2
col <- colorRampPalette(c("green","white","red"))(256)
pheatmap(deg_expr,col=col,fontsize=8,show_colnames =F,show_rownames = F,
filename = paste0("figures/",filename,".png"))
return(paste0("figures/",filename,".png"))
}
Volcano <-function(deg,filename,title){
# 火山图----------------------
getDeg(deg)%>%
mutate(direction = factor(ifelse(Pvalue < 0.01 & abs(logFC)>2,
ifelse(logFC>1,"Up","Down"),
"NS"),levels = c("Up","Down","NS"))) %>%
na.omit()%>%
ggplot(aes(x=logFC,y=-log10(Pvalue),colour=direction))+
geom_point(alpha=0.6) +
scale_color_manual(values = c("red","green","#000000"))+
xlab(expression(log[2]("Fold Change")))+
ylab(expression(-log[10]("P Value")))+
theme_bw()+
theme(legend.title = element_blank(),
plot.title = element_text(hjust = 0.5,size = 24, face= "bold"),
axis.title = element_text(size=20),
legend.text= element_text(size=20),
aspect.ratio=1)+
geom_vline(xintercept = c(-2,2),lty=2,col="black",lwd=0.6)+
geom_hline(yintercept = -log10(0.01),lty=2,col="black",lwd=0.6) +
labs(title = title)+
geom_text_repel(data=. %>%filter(Pvalue<0.001,abs(logFC)>2),
aes(label=symbol),
size=3,
segment.color="black",
show.legend = F)+
ggsave(filename = paste0("figures/",filename,".png"),width = 20,height = 15,units="cm")
return(paste0("figures/",filename,".png"))
}
get_mircode <- function(){
if(!file.exists("result/mircode.rda")){
mircode <- read.table("/home/wangyang/workspace/bioinfo_analysis/Rscript/data/mircode_highconsfamilies.txt",sep = "\t",header = T)
table(mircode$gene_class)
lncmiRcode = mircode[mircode$gene_class %in%c("lncRNA, intergenic","lncRNA, overlapping"),1:4]
head(lncmiRcode$microrna)
library(stringr)
p1 = str_starts(lncmiRcode$microrna,"miR-")
table(p1)
p2 = str_starts(lncmiRcode$microrna,"let-")
table(p2)
lncmiRcode_let <- lncmiRcode[p2,]%>%
dplyr::select(c("gene_symbol","microrna"))%>%
mutate(miRNA=str_replace_all(microrna,"let-",""))%>%
separate_rows(miRNA,sep = "/",convert = T)%>%
mutate_at(vars(contains("miRNA")), ~ paste0("hsa-let-",.))%>%
mutate(miRNA = str_extract(miRNA,"hsa-let-[0-9]+[a-z]?"))%>%
dplyr::select(c("gene_symbol","miRNA"))
lncmiRcode_miR <- lncmiRcode[p1,]%>%
dplyr::select(c("gene_symbol","microrna"))%>%
mutate(miRNA=str_replace_all(microrna,"miR-",""))%>%
separate_rows(miRNA,sep = "/",convert = T)%>%
mutate_at(vars(contains("miRNA")), ~ paste0("hsa-mir-",.))%>%
mutate(miRNA = str_extract(miRNA,"hsa-mir-[0-9]+[a-z]?"))%>%
dplyr::select(c("gene_symbol","miRNA"))
lncmiRcode_f <- rbind(lncmiRcode_let,lncmiRcode_miR)
saveRDS(lncmiRcode_f,file = "result/mircode.rda")
return(lncmiRcode_f)
}
return(readRDS("result/mircode.rda"))
}
Volcano_sig <-function(deg,title=NULL,filename=NULL,gene=NULL){
# 火山图----------------------
res <- getDeg(deg)%>%
mutate(direction = factor(ifelse(Pvalue < 0.01 & abs(logFC)>2,
ifelse(logFC>1,"Up","Down"),
"NS"),levels = c("Up","Down","NS"))) %>%
na.omit()%>%
ggplot(aes(x=logFC,y=-log10(Pvalue),colour=direction))+
geom_point(alpha=0.6) +
scale_color_manual(values = c("red","green","#000000"))+
xlab(expression(log[2]("Fold Change")))+
ylab(expression(-log[10]("P Value")))+
theme_bw()+
theme(legend.title = element_blank(),
plot.title = element_text(hjust = 0.5,size = 24, face= "bold"),
axis.title = element_text(size=20),
legend.text= element_text(size=20),
aspect.ratio=1)+
geom_vline(xintercept = c(-2,2),lty=2,col="black",lwd=0.6)+
geom_hline(yintercept = -log10(0.01),lty=2,col="black",lwd=0.6)
if(!is.null(title)){
res <- res+labs(title = title)
}
if(!is.null(gene)){
res <- res + geom_text_repel(data=. %>%filter(symbol %in% gene),
aes(label=symbol),
size=3,
segment.color="black",
show.legend = F)
}
if(!is.null(filename)){
res <- res + ggsave(filename = paste0("figures/",filename,".png"),width = 20,height = 15,units="cm")
}
return(res)
}
basesave <- function(expr, filename, print=T) {
#extension
exten = stringr::str_match(filename, "\\.(\\w+)$")[, 2]
switch(exten,
png = {
png(filename)
eval(expr, envir = parent.frame())
dev.off()
},
{stop("filetype not recognized")})
#print?
if (print) eval(expr, envir = parent.frame())
invisible(NULL)
}
}
#####################################################
## 热图火山图
#####################################################
(function(){
deg <- readRDS("result/limma_mRNA_deg.rds")
Volcano(deg,"GEO_Volcano_heatmap/1_mRNA_Volcano",title = "mRNA Volcano")
expr <- readRDS("result/gset_expr_annot_mRNA_GSE89102.rda")
Pheatmap(deg,expr,filename="GEO_Volcano_heatmap/1_mRNA_pheatmap")
deg <- readRDS("result/limma_lnRNA_deg.rds")
Volcano(deg,"GEO_Volcano_heatmap/1_lnRNA_Volcano",title = "lnRNA Volcano")
expr <- readRDS("result/gset_expr_annot_lnRNA_GSE89102.rda")
Pheatmap(deg,expr,filename="GEO_Volcano_heatmap/1_lnRNA_pheatmap")
deg <- readRDS("result/limma_miRNA_deg_2.rds")
Volcano(deg,"GEO_Volcano_heatmap/1_miRNA_Volcano",title = "miRNA Volcano")
if(F){
res <- getDeg(deg)%>%
mutate(direction = factor(ifelse(Pvalue < 0.05 & abs(logFC)>2,
ifelse(logFC>1,"Up","Down"),
"NS"),levels = c("Up","Down","NS"))) %>%
na.omit()%>%
ggplot(aes(x=logFC,y=-log10(Pvalue),colour=direction))+
geom_point(alpha=0.6) +
scale_color_manual(values = c("red","green","#000000"))+
xlab(expression(log[2]("Fold Change")))+
ylab(expression(-log[10]("P Value")))+
theme_bw()+
theme(legend.title = element_blank(),
plot.title = element_text(hjust = 0.5,size = 24, face= "bold"),
axis.title = element_text(size=20),
legend.text= element_text(size=20),
aspect.ratio=1)+
geom_vline(xintercept = c(-2,2),lty=2,col="black",lwd=0.6)+
geom_hline(yintercept = -log10(0.05),lty=2,col="black",lwd=0.6)
print(res)
}
expr <- readRDS("result/gset_expr_annot_miRNA_GSE59973.rda")
Pheatmap(deg,expr,filename="GEO_Volcano_heatmap/1_miRNA_pheatmap")
if(F){
deg_gene <- getDeg(deg)%>%
filter(abs(logFC)>2,Pvalue<0.05)%>%
arrange(desc(abs(logFC)))%>%
pull("symbol")
deg_expr <- expr[deg_gene,]
#deg_expr <- t(scale(t(deg_expr)))
#deg_expr[deg_expr > 2] = 2
#deg_expr[deg_expr < -2] = -2
col <- colorRampPalette(c("green","white","red"))(256)
pheatmap(deg_expr,col=col,fontsize=8,show_colnames =F,show_rownames = F)
# return(paste0("figures/",filename,".png"))
}
})()
if(F){
deg <- readRDS("result/miRNA_iso_expr_DESeq.rda")
Volcano(deg,"1_miRNA_iso_Volcano",title = "miRNA Volcano")
expr <- readRDS("result/miRNA_iso_expr.rda")
Pheatmap(deg,expr,filename="1_miRNA_iso_pheatmap")
deg_gene <- getDeg(deg)%>%
filter(abs(logFC)>1,Pvalue<0.05)%>%
arrange(desc(abs(logFC)))%>%
pull("symbol")
deg_expr <- expr[deg_gene,]%>%
{log10(cpm(.)+0.01)}
#deg_expr <- t(scale(t(deg_expr)))
#deg_expr[deg_expr > 2] = 2
#deg_expr[deg_expr < -2] = -2
col <- colorRampPalette(c("green","white","red"))(256)
pheatmap(deg_expr,col=col,fontsize=8,show_colnames =F,show_rownames = F,
filename = "figures//1_miRNA_iso_pheatmap.png")
}
#############################################
## 差异基因提取 变量
#############################################
miRNA_deg_sig <- getDeg(readRDS("result/limma_miRNA_deg_2.rda"))%>%
filter(abs(logFC)>2,Pvalue<0.01)%>%
mutate(symbol=sub("R","r",symbol))%>%
mutate(symbol= case_when(grepl("hsa-mir",symbol)~str_extract(symbol,"hsa-mir-[0-9]+[a-z]?"),
grepl("hsa-let",symbol)~str_extract(symbol,"hsa-let-[0-9]+[a-z]?")))%>%
arrange(Pvalue)
report("以abs(logFC)>2 & Pvalue<0.01 为条件筛选差异miRNA共 ",length(unique(miRNA_deg_sig$symbol))," 个")
lncRNA_deg_sig <- getDeg(readRDS("result/limma_lnRNA_deg.rda"))%>%
filter(abs(logFC)>2,Pvalue<0.01)
cat("以abs(logFC)>2 & Pvalue<0.01 为条件筛选差异lncRNA共 ",length(unique(lncRNA_deg_sig$symbol))," 个")
mRNA_deg_sig <- getDeg(readRDS("result/limma_mRNA_deg.rda"))%>%
filter(abs(logFC)>2,Pvalue<0.01)
cat("以abs(logFC)>2 & Pvalue<0.01 为条件筛选差异mRNA共 ",length(unique(mRNA_deg_sig$symbol))," 个")
lnRNA_expr <- readRDS("result/lncRNA_expr.rda")%>%
dplyr::select(!matches("TCGA-([A-Z 0-9]*?)-([A-Z 0-9]*?)-(11.*?)-(.*?)"))%>%
rownames_to_column("symbol")%>%
mutate(symbol=str_extract(symbol,"[A-Z 0-9 -]+"))
lnRNA_expr <- lnRNA_expr[!duplicated(lnRNA_expr$symbol),]%>%
rownames_to_column("id")%>%
select(-id)%>%
column_to_rownames("symbol")
sum(rownames(lnRNA_expr) %in% lncRNA_deg_sig$symbol)
lnRNA_expr <- lnRNA_expr[rownames(lnRNA_expr) %in% lncRNA_deg_sig$symbol,]
mRNA_expr <- readRDS("result/mRNA_expr.rda")%>%
dplyr::select(!matches("TCGA-([A-Z 0-9]*?)-([A-Z 0-9]*?)-(11.*?)-(.*?)"))
miRNA_expr <- readRDS("result/miRNA_expr.rda")%>%
dplyr::select(!matches("TCGA-([A-Z 0-9]*?)-([A-Z 0-9]*?)-(11.*?)-(.*?)"))%>%
rownames_to_column("symbol")%>%
mutate(symbol= case_when(grepl("hsa-mir",symbol)~str_extract(symbol,"hsa-mir-[0-9]+[a-z]?"),
grepl("hsa-let",symbol)~str_extract(symbol,"hsa-let-[0-9]+[a-z]?")))%>%
aggregate(.~symbol,.,mean)%>%
column_to_rownames("symbol")
#############################################
## lnRNA ralated miRNA
#############################################
## 获得miRNA与lnRNA对应关系
(function(){
cat("\n***************************lnRNA ralated miRNA***************************")
if(T){
mircode <- read_tsv("/home/wangyang/workspace/pipeline/CANCERjupyter/Common/lncRNA_miRCODE_miRNA.csv")%>%
plyr::rename(c(lncRNA="gene_symbol"))
}else{
mircode <- get_mircode()
}
cat("从miRcode数据库获得lnRNA-miRNA的对应关系:共有 ",dim(mircode)[1]," 个")
lnRNAPrediction <- mircode%>%
filter(gene_symbol %in% rownames(lnRNA_expr))
cat("在miRcode中找到的lnRNA共有 ",length(unique(lnRNAPrediction$gene_symbol))," 个,","对应的miRNA有 ",length(unique(lnRNAPrediction$miRNA))," 个")
miRNA_lnRNA <- lnRNAPrediction%>%
filter(miRNA %in% miRNA_deg_sig$symbol)%>%
plyr::rename(c(gene_symbol="lnRNA"))%>%
dplyr::select(-gene_id)
cat("预测得到的 ",length(unique(lnRNAPrediction$miRNA))," 个miRNA与差异的miRNA取交集, 得到 ",length(unique(miRNA_lnRNA$miRNA)), "个miRNA")
miRNA_lnRNA <- miRNA_lnRNA[!duplicated(miRNA_lnRNA),]
saveRDS(miRNA_lnRNA,file = "result/miRNA_lnRNA.rda")
miRNA_intersect <- unique(miRNA_lnRNA$miRNA)
lnRNA_intersect <- unique(miRNA_lnRNA$lnRNA)
#length(miRNA_intersect)
#length(lnRNA_intersect)
cat("最后得到 ",length(lnRNA_intersect)," 个lncRNA可以与 ",length(miRNA_intersect)," 个miRNA相互作用")
#png(filename="plot_%02d.png")
png(file="figures/venn/ggvenn_lnRNA_prediction_miRNA.png",
width = 15,height = 15,units = "cm",bg = "white",res=300)
res <- ggvenn(
list("lncRNA-ralated miRNA"=lnRNAPrediction$miRNA,"DE miRNAs"=miRNA_deg_sig$symbol),
stroke_size = 0.5, set_name_size = 5,text_size = 5
)
print(res)
dev.off()
cat("lnRNA预测miRNA与差异miRNA Venn 图见figures/ggvenn_lnRNA_prediction_miRNA.png")
cat("***************************lnRNA ralated miRNA***************************")
})()
#############################################
## miRNA ralated mRNA
#############################################
(function(){
cat("\n***************************miRNA ralated mRNA***************************")
miRNA_lnRNA <- readRDS("result/miRNA_lnRNA.rda")
miRNA_intersect <- unique(miRNA_lnRNA$miRNA)
## test start hsa-let-7a-2 预测时去掉 -2
if(F){
miRNAprediction <- SpidermiRdownload_miRNAprediction(mirna_list=c("hsa-let-7a"))
}
## test end
if(!file.exists("result/miRNAprediction.rda")){
miRNAprediction <- SpidermiRdownload_miRNAprediction(mirna_list=miRNA_intersect)
save(miRNAprediction,file = "result/miRNAprediction.rda")
}else{
load("result/miRNAprediction.rda")
}
cat("用 ",length(miRNA_intersect)," 个miRMA使用[R package SpidermiR]预测得到 ",length(miRNAprediction$V2)," 个mRNA")
miRNA_mRNA <- miRNAprediction%>%
plyr::rename(c(V1="miRNA",V2="mRNA"))%>%
filter(mRNA %in% mRNA_deg_sig$symbol)
miRNA_mRNA <- miRNA_mRNA[!duplicated(miRNA_mRNA),]
saveRDS(miRNA_mRNA,file = "result/miRNA_mRNA.rda")
cat("用预测得到mRNA与差异的miRNA去交集, 得到 ",length(unique(miRNA_mRNA$mRNA)), "个mRNA")
miRNA_intersect <- unique(miRNA_mRNA$miRNA)
mRNA_intersect <- unique(miRNA_mRNA$mRNA)
#length(miRNA_intersect)
#length(mRNA_intersect)
cat("最后得到 ",length(miRNA_intersect)," 个miRNA可以靶向 ",length(mRNA_intersect)," 个mRNA相互作用")
library("ggvenn")
png(file="figures/venn/ggvenn_miRNA_predication_mRNA.png",
width = 15,height = 15,units = "cm",bg = "white",res=300)
res <- ggvenn(
list("miRNA-ralated mRNA"=miRNAprediction$V2,"DE mRNAs"=mRNA_deg_sig$symbol),
stroke_size = 0.5, set_name_size = 5,text_size = 5
)
print(res)
dev.off()
cat("miRNA预测mRNA与差异miRNA Venn 图见figures/ggvenn_miRNA_predication_mRNA.png")
cat("***************************lnRNA ralated miRNA***************************")
})()
if(F){
list<-SpidermiRdownload_miRNAvalidate(validated)
list%>%
filter(V1 %in% miRNA_intersect)
org<-SpidermiRquery_species(species)
net_type<-SpidermiRquery_networks_type(organismID=org[6,])
net_shar_prot<-SpidermiRquery_spec_networks(organismID = org[6,]
,network = "pred")
}
#############################################
### 构建ceRNA网络
#############################################
if(F){
getDeg(readRDS("result/miRNA_expr_DESeq.rda"))%>%
mutate(direction = factor(ifelse(Pvalue < 0.01 & abs(logFC)>2,
ifelse(logFC>1,"Up","Down"),
"NS"),levels = c("Up","Down","NS"))) %>%
na.omit()%>%
ggplot(aes(x=logFC,y=-log10(Pvalue),colour=direction))+
geom_point(alpha=0.6) +
scale_color_manual(values = c("red","green","#000000"))+
xlab(expression(log[2]("Fold Change")))+
ylab(expression(-log[10]("P Value")))+
theme_bw()+
theme(legend.title = element_blank(),
plot.title = element_text(hjust = 0.5,size = 24, face= "bold"),
axis.title = element_text(size=20),
legend.text= element_text(size=20),
aspect.ratio=1)+
geom_vline(xintercept = c(-2,2),lty=2,col="black",lwd=0.6)+
geom_hline(yintercept = -log10(0.01),lty=2,col="black",lwd=0.6) +
geom_text_repel(data=. %>%filter(symbol %in% c("hsa-mir-301b")),
aes(label=symbol),
size=3,
segment.color="black",
show.legend = F)
}
report("\n***************************构建ceRNA网络***************************")
miRNA_lnRNA <- readRDS("result/miRNA_lnRNA.rda")
miRNA_mRNA <- readRDS("result/miRNA_mRNA.rda")
lnRNA_miRNA_mRNA <- merge(miRNA_lnRNA,miRNA_mRNA,by="miRNA")
if(F){
miRNA <- unique(lnRNA_miRNA_mRNA$miRNA)
length(miRNA)
#Volcano_sig(readRDS("result/miRNA_expr_DESeq.rda"),gene=miRNA)
lnRNA <- unique(lnRNA_miRNA_mRNA$lnRNA)
length(lnRNA)
#Volcano_sig(readRDS("result/lnRNA_expr_DESeq.rda"),gene=lnRNA)
mRNA <-unique(lnRNA_miRNA_mRNA$mRNA)
length(mRNA)
}
if(T){
lncRNA_deg_ce <- lncRNA_deg_sig%>%
plyr::rename(c(symbol="lnRNA"))%>%
mutate(lnRNA_direction=ifelse(logFC>0,"up","down"))%>%
dplyr::select(lnRNA,lnRNA_direction)
miRNA_deg_ce <- miRNA_deg_sig%>%
plyr::rename(c(symbol="miRNA"))%>%
mutate(miRNA_direction=ifelse(logFC>0,"up","down"))%>%
dplyr::select(miRNA,miRNA_direction)
mRNA_deg_ce <- mRNA_deg_sig%>%
plyr::rename(c(symbol="mRNA"))%>%
mutate(mRNA_direction=ifelse(logFC>0,"up","down"))%>%
dplyr::select(mRNA,mRNA_direction)
lnRNA_miRNA_mRNA_ <- lnRNA_miRNA_mRNA
lnRNA_miRNA_mRNA <- lnRNA_miRNA_mRNA_ %>%
inner_join(lncRNA_deg_ce,by="lnRNA")%>%
inner_join(miRNA_deg_ce,by="miRNA")%>%
inner_join(mRNA_deg_ce,by="mRNA")%>%
filter(lnRNA_direction!=miRNA_direction)%>%
filter(miRNA_direction!=mRNA_direction)
}
#saveRDS(lnRNA_miRNA_mRNA,file = "result/lnRNA_miRNA_mRNA.rda")
lnRNA_miRNA_mRNA <- readRDS("result/lnRNA_miRNA_mRNA.rda")
unique(lnRNA_miRNA_mRNA$miRNA)
length(unique(lnRNA_miRNA_mRNA$lnRNA))
ce_lnRNA <- unique(lnRNA_miRNA_mRNA$lnRNA)
ce_lnRNA <- ce_lnRNA[ce_lnRNA %in% rownames(lnRNA_expr)]
ce_miRNA <- unique(lnRNA_miRNA_mRNA$miRNA)
ce_miRNA <- ce_lnRNA[ce_miRNA %in% rownames(mRNA_expr)]
ce_mRNA <- unique(lnRNA_miRNA_mRNA$mRNA)
ce_mRNA <- ce_lnRNA[ce_miRNA %in% rownames(mRNA_expr)]
cytoscape <- function(lnRNA_miRNA_mRNA,filename){
ce_lnRNA <- unique(lnRNA_miRNA_mRNA$lnRNA)
ce_miRNA <- unique(lnRNA_miRNA_mRNA$miRNA)
ce_mRNA <- unique(lnRNA_miRNA_mRNA$mRNA)
report("ceRNA网络中有: ", length(ce_lnRNA),
" 个lnRNA对应 ",length(ce_miRNA)," 个miRNA, ",
length(ce_miRNA)," 的miRNA对应 ",
length(ce_mRNA), "的mRNA")
miRNA_lnRNA <<- unique(lnRNA_miRNA_mRNA%>%dplyr::select(lnRNA,miRNA))
miRNA_mRNA <<- unique(lnRNA_miRNA_mRNA%>%dplyr::select(miRNA,mRNA))
lncRNA_deg_ce <- lncRNA_deg_sig%>%
plyr::rename(c(symbol="lnRNA"))%>%
filter(lnRNA %in% ce_lnRNA)%>%
mutate(direction=ifelse(logFC>0,"up","down"),type="lncRNA")%>%
dplyr::select(name=lnRNA,direction,type)
miRNA_deg_ce <- miRNA_deg_sig%>%
plyr::rename(c(symbol="miRNA"))%>%
filter(miRNA %in% ce_miRNA)%>%
mutate(direction=ifelse(logFC>0,"up","down"),type="miRNA")%>%
dplyr::select(name=miRNA,direction,type)
mRNA_deg_ce <- mRNA_deg_sig%>%
plyr::rename(c(symbol="mRNA"))%>%
filter(mRNA %in% ce_mRNA)%>%
mutate(direction=ifelse(logFC>0,"up","down"),type="mRNA")%>%
dplyr::select(name=mRNA,direction,type)
cytoscape_type <- bind_rows(mRNA_deg_ce,miRNA_deg_ce,lncRNA_deg_ce)%>%
mutate(type = str_c(direction,type,sep="_"))%>%
dplyr::select(-2)
write.csv(cytoscape_type,file = paste0("result/",filename,"_type.csv"),row.names = F,quote = F)
report("写入网络节点类型到 ",paste0("result/",filename,"_type.csv"), " 共有 ",dim(cytoscape_type)[1]," 个")
ceRAN_pair1 <- miRNA_lnRNA%>%
dplyr::select(miRNA,name=lnRNA)
ceRAN_pair2 <- miRNA_mRNA%>%
dplyr::select(miRNA,name=mRNA)
cytoscape_input <- bind_rows(ceRAN_pair1,ceRAN_pair2)
write.csv(cytoscape_input,file = paste0("result/",filename,"_input.csv"),row.names = F,quote = F)
report("写入网络节点关系:",paste0("result/",filename,"_input.csv")," 共有 ",dim(cytoscape_input)[1]," 个")
}
cytoscape(lnRNA_miRNA_mRNA,filename = "geo__")
a <- unique(lnRNA_miRNA_mRNA$mRNA)
length(a)
write.csv(a,file = "result/gene_all.csv",quote = F,row.names = F)
genes <- "MDM2,CCN1,HIF1A,TGFB1,MYCN,MXD1,NSD3,BTG1,PPP3R1,PPP6C"
genes <- unlist(str_split(genes,pattern = ","))
lnRNA_miRNA_mRNA_top10 <- lnRNA_miRNA_mRNA%>%
filter(mRNA %in% genes)
cytoscape(lnRNA_miRNA_mRNA_top10,filename = "geo_top10")
genes %in% unique(lnRNA_miRNA_mRNA_top10$mRNA)
unique(lnRNA_miRNA_mRNA_top10$mRNA)
unique(lnRNA_miRNA_mRNA_top10$lnRNA)
#Volcano_sig(readRDS("result/mRNA_expr_DESeq.rda"),gene=mRNA)
report("***************************构建ceRNA网络***************************")
#############################################
## lncRNA miRNA mRNA 生存分析
#############################################
report("\n***************************lncRNA miRNA mRNA 生存分析***************************")
library(survival)
library(survminer)
survival_p <- function(data,gene,prefix,pcutoff=0.05){
low_high <- ifelse(data[,gene]<=median(data[,gene]),"Low","Hight")
diff <- survdiff(Surv(futime_year,fustat)~low_high,data = data)
pVal <- 1 -pchisq(diff$chisq,df =1)
pValue <- signif(pVal,4)
if(pValue<pcutoff){
fit <- surv_fit(Surv(futime_year,fustat) ~ low_high, data = data)
ggsurvplot(fit, pval=T, risk.table=F,
legend.title=gene,
risk.table.height = 0.3,data = data)+
ggsave(filename = paste0("figures/survival/",prefix,"-",gene,".png"))
}
return(pValue)
}
Survival <- function(expr,gene,clinical,prefix,pcutoff=0.05){
expr_survival <- expr[gene,]%>%
t()%>%
as.data.frame()%>%
rownames_to_column("TCGA_full_id")%>%
mutate(patient_id =substr(TCGA_full_id,1,12))%>%
inner_join(clinical,by="patient_id")
mRNA_survival <- sapply(gene,
function(x){ survival_p(expr_survival,x,prefix,pcutoff)})
return(mRNA_survival)
}
mRNA_lnRNA_clinical <- readRDS("result/mRNA_lnRNA_clinical.rda")
res <- Survival(mRNA_expr,ce_mRNA,mRNA_lnRNA_clinical,prefix="mRNA")
report("使用[R pacakge Survival]得到 ",length(ce_mRNA)," 个mRNA中(以p<0.05筛选)有 ",sum(res<0.05)," 个mRNA与总生存率相关")
report(paste(names(res[res<0.05]),collapse = ","))
## test start ceRNAw网络mRNA的表达与患者总生存率之间关系
if(F){
mRNA_expr_survival <- mRNA_expr[mRNA,]%>%
t()%>%
as.data.frame()%>%
rownames_to_column("TCGA_full_id")%>%
mutate(patient_id =substr(TCGA_full_id,1,12))%>%
inner_join(mRNA_lnRNA_clinical,by="patient_id")
survival_p(mRNA_expr_survival,"PHOX2B")
mRNA_survival <- sapply(mRNA,
function(x){ survival_p(mRNA_expr_survival,x)})
table(mRNA_survival<0.05)
mRNA_expr_survival <- mRNA_expr_survival%>% filter(futime_year!=0)
low_high <- ifelse(mRNA_expr_survival[,"PHOX2B"] <= median(mRNA_expr_survival[,"PHOX2B"]),"Low","Hight")
mRNA_expr_survival$futime_year <- as.numeric(mRNA_expr_survival$futime_year)
mRNA_expr_survival$fustat <- as.numeric(mRNA_expr_survival$fustat)
fit <- surv_fit(Surv(futime_year,fustat) ~ low_high, data = mRNA_expr_survival)
# pdf(file = paste0("result/PHOX2B.pdf"),width = 5.5,height = 5)
ggsurvplot(fit, pval=T, risk.table=F,
legend.title='PHOX2B',
risk.table.height = 0.3,data = mRNA_expr_survival)+
ggsave(filename = "result/aa.png")
# dev.off()
# ggsurvplot(fit, data = mRNA_expr_survival,
# surv.median.line = "hv", # 增加中位生存时间
# conf.int = TRUE) # 增加置信区间
}
## test End
mRNA_lnRNA_clinical <- readRDS("result/mRNA_lnRNA_clinical.rda")
res <- Survival(lnRNA_expr,ce_lnRNA,mRNA_lnRNA_clinical,prefix="lnRNA")
report("使用[R pacakge Survival]得到 ",length(ce_lnRNA)," 个lnRNA中(以p<0.05筛选)有 ",sum(res<0.05)," 个lnRNA与总生存率相关")
report(paste(names(res[res<0.05]),collapse = ","))
## 由于miRNA存在hsa-let-7a-2 这种形式,预测mRNA时是去掉-2 ,以hsa-let-7a预测
miRNA_clinical <- readRDS("result/miRNA_clinical.rda")
res <- Survival(miRNA_expr,ce_miRNA,miRNA_clinical,prefix="miRNA",pcutoff = 0.09)
report("使用[R pacakge Survival]得到 ",length(ce_miRNA)," 个lnRNA中(以p<0.05筛选)有 ",sum(res<0.09)," 个lnRNA与总生存率相关")
report(paste(names(res[res<0.09]),collapse = ","))
report("***************************lncRNA miRNA mRNA 生存分析***************************")
#############################################
## cox 回归分析
#############################################
library(survivalROC)
library(survival)
library(survminer)
Cox_single <- function(data,gene,filename=NULL){
outTab <- data.frame()
for(i in gene){
cox <- coxph(Surv(futime_year,fustat) ~ data[,i],data=data)
coxSummary <- summary(cox)
outTab <- rbind(outTab,cbind(id=i,
HR=coxSummary$conf.int[,"exp(coef)"],
HR.95L=coxSummary$conf.int[,"lower .95"],
HR.95H=coxSummary$conf.int[,"upper .95"],
pvalue=coxSummary$coefficients[,"Pr(>|z|)"]))
}
if(!is.null(filename)){
write.csv(outTab,file = paste0("result/",filename,".csv"),quote = F,row.names = F)
}
return(outTab)
}
Cox_multi <- function(data,gene,filename=NULL){
if(sum(grepl("-",gene))>0){
gene <- sapply(gene,function(x){sub("-","",x)})
data<- data%>%
rename_all(~sub("-","",.))
}
formula <-as.formula(paste0("Surv(futime_year,fustat)~",paste0(gene,collapse = "+")))
cox <- coxph(formula,data=data)
#cox <- step(cox,direction = "both")
coxSummary <- summary(cox)
outTab <- data.frame()
outTab <- rbind(outTab,cbind(HR=coxSummary$conf.int[,"exp(coef)"],
HR.95L=coxSummary$conf.int[,"lower .95"],
HR.95H=coxSummary$conf.int[,"upper .95"],
pvalue=coxSummary$coefficients[,"Pr(>|z|)"]))
riskScore <- predict(cox,type = "risk",newdata = data)
expr_survival_riskScore <- data %>%
mutate(riskScore = as.vector(ifelse(riskScore>median(riskScore),"high","low")))
fit <- surv_fit(Surv(futime_year,fustat) ~ riskScore, data = expr_survival_riskScore)
res <- ggsurvplot(fit, pval=T, risk.table=F,
risk.table.height = 0.3,data = expr_survival_riskScore)
if(!is.null(filename)){
res+ ggsave(filename = paste0("figures/cox/",filename,".png"))
write.csv(outTab,file = paste0("result/",filename,".csv"),quote = F,row.names = F)
}
return(list(outTab=outTab,image=res))
}
riskScore_pheatmap <- function(data,gene,filename=NULL){
if(sum(grepl("-",gene))>0){
gene <- sapply(gene,function(x){sub("-","",x)})
data<- lnRNA_expr_survival%>%
rename_all(~sub("-","",.))
}
formula <-as.formula(paste0("Surv(futime_year,fustat)~",paste0(gene,collapse = "+")))
cox <- coxph(formula,data=data)
#cox <- step(cox,direction = "both")
riskScore <- predict(cox,type = "risk",newdata = data)
expr_survival_riskScore <- data %>%
mutate(riskScore = as.vector(ifelse(riskScore>median(riskScore),"high","low")))
deg_expr <- expr[deg_gene,]%>%
{log10(cpm(.)+0.01)}
#deg_expr <- t(scale(t(deg_expr)))
#deg_expr[deg_expr > 2] = 2
#deg_expr[deg_expr < -2] = -2
col <- colorRampPalette(c("green","white","red"))(256)
pheatmap(deg_expr,col=col,fontsize=8,show_colnames =F,show_rownames = F,
filename = paste0("figures/cox/",filename,".png"))
return(paste0("figures/",filename,".png"))
}
Cox_multi_ROC <- function(data,gene,filename=NULL,predict.time=5){
if(sum(grepl("-",gene))>0){
gene <- sapply(gene,function(x){sub("-","",x)})
data<- data%>%
rename_all(~sub("-","",.))
}
formula <-as.formula(paste0("Surv(futime_year,fustat)~",paste0(gene,collapse = "+")))
cox <- coxph(formula,data=data)
#message(summary(cox))
#cox <- step(cox,direction = "both")
riskScore <- predict(cox,type = "risk",newdata = data)
roc <- survivalROC(Stime=data$futime_year ,status=data$fustat,marker=riskScore,
predict.time=predict.time,method="KM")
res <- xyplot(TP~FP,roc,main=paste("ROC curve(","AUC=",round(roc$AUC,3),")"),
xlab="False postive rate",ylab="True positive rate",panel = function(x,y){
panel.xyplot(x,y,type="l",lwd=2,cex.main=1.3,cex.lab=1.2,cex.axis=1.2,font=1.2,col="red")
panel.abline(c(0,1))
})
if(!is.null(filename)){
png(filename = paste0("figures/cox/",filename,".png"))
print(res)
dev.off()
}
return(res)
}
lnRNA_expr <- readRDS("result/lncRNA_expr.rda")
mRNA_lnRNA_clinical <- readRDS("result/mRNA_lnRNA_clinical.rda")
lnRNA_expr_survival <- lnRNA_expr[lnRNA_intersect,]%>%
filter()%>%
t()%>%
as.data.frame()%>%
rownames_to_column("TCGA_full_id")%>%
mutate(patient_id =substr(TCGA_full_id,1,12))%>%
inner_join(mRNA_lnRNA_clinical,by="patient_id")
if(F){
lnRNA_expr_ <- lnRNA_expr[ce_lnRNA,]
#lnRNA_expr_ <- log2(lnRNA_expr_+0.01)
par(mfcol=c(2,1))
boxplot(lnRNA_expr_)
boxplot(log2(lnRNA_expr_+1))
boxplot( log2(lnRNA_expr_+0.01))
boxplot( log2(lnRNA_expr_+1))
boxplot(scale(cpm(lnRNA_expr_),center = F, scale = TRUE))
lnRNA_expr_survival <- lnRNA_expr_%>%
t()%>%
as.data.frame()%>%
rownames_to_column("TCGA_full_id")%>%
mutate(patient_id =substr(TCGA_full_id,1,12))%>%
inner_join(mRNA_lnRNA_clinical,by="patient_id")
Cox_single_res <- Cox_single(lnRNA_expr_survival,ce_lnRNA,filename = "singleCox_lnRNA")
genes <- Cox_single_res%>%filter(pvalue<0.09)%>%pull("id")
Cox_multi(lnRNA_expr_survival,genes)$image
Cox_multi_ROC(lnRNA_expr_survival,genes,predict.time=3)
}
## 22个lnRNA 先单因素cox再多因素cox LINC00486,LINC00365
Cox_single_res <- Cox_single(lnRNA_expr_survival,lnRNA_intersect,filename = "singleCox_lnRNA")
genes <- Cox_single_res%>%filter(pvalue<0.09)%>%pull("id")
saveRDS(genes,file = "result/lnRNA_cox_mutil.rda")
res <- Cox_multi(lnRNA_expr_survival,genes,filename = "muitiCox_lnRNA")
res$outTab
print(res$image)
Cox_multi_ROC(lnRNA_expr_survival,genes,predict.time=3,filename = "muitiCox_lnRNA_lnRNA_ROC")
if(F){
cox <- coxph(Surv(futime_year,fustat) ~ lnRNA_expr_survival[,"HOTAIR"],data=lnRNA_expr_survival)
coxSummary <- summary(cox)
outTab <- data.frame()
outTab <- rbind(outTab,cbind(HR=coxSummary$conf.int[,"exp(coef)"],
HR.95L=coxSummary$conf.int[,"lower .95"],
HR.95H=coxSummary$conf.int[,"upper .95"],
pvalue=coxSummary$coefficients[,"Pr(>|z|)"]))
wald.test <- signif(coxSummary$wald["pvalue"],digits = 3)
beta <- signif(coxSummary$wald["test"],digits = 3)
HR <- signif(coxSummary$coef[1],digits = 3)
HR.lower = coxSummary$conf.int[,"lower .95"]
HR.upper = coxSummary$conf.int[,"upper .95"]
}
if(F){
if(sum(grepl("-",genes))>0){
genes <- sapply(genes,function(x){sub("-","",x)})
lnRNA_expr_survival<- lnRNA_expr_survival%>%
rename_all(~sub("-","",.))
}
formula <-as.formula(paste0("Surv(futime_year,fustat)~",paste0(genes,collapse = "+")))
cox <- coxph(formula,data=lnRNA_expr_survival)
cox <- step(cox,direction = "both")
coxSummary <- summary(cox)
outTab <- data.frame()
outTab <- rbind(outTab,cbind(HR=coxSummary$conf.int[,"exp(coef)"],
HR.95L=coxSummary$conf.int[,"lower .95"],
HR.95H=coxSummary$conf.int[,"upper .95"],
pvalue=coxSummary$coefficients[,"Pr(>|z|)"]))
riskScore <- predict(cox,type = "risk",newdata = lnRNA_expr_survival)
lnRNA_expr_survival_riskScore <- lnRNA_expr_survival %>%
mutate(riskScore = as.vector(ifelse(riskScore< median(riskScore),"high","low")))
fit <- surv_fit(Surv(futime_year,fustat) ~ riskScore, data = lnRNA_expr_survival_riskScore)
ggsurvplot(fit, pval=T, risk.table=F,
risk.table.height = 0.3,data = lnRNA_expr_survival_riskScore,xlab="Time(Years)")
roc <- survivalROC(Stime=lnRNA_expr_survival_riskScore$futime_year ,status=lnRNA_expr_survival_riskScore$fustat,marker=riskScore,
predict.time=5,method="KM")
plot(roc$FP,roc$TP,type="l",xlim=c(0,1),ylim=c(0,1),col="red",main=paste("ROC curve(","AUC=",round(roc$AUC,3),")"),
xlab="False postive rate",ylab="True positive rate",
lwd=2,cex.main=1.3,cex.lab=1.2,cex.axis=1.2,font=1.2)
abline(0,1)
lnRNA_expr_survival <- lnRNA_expr_survival%>%
select(c(genes,futime_year,fustat))
library(ggrisk)
ggrisk(lnRNA_expr_survival,time="futime_year",event="fustat",heatmap.genes = genes)
}
if(F){
formula <-as.formula(paste0("Surv(futime_year,fustat)~",paste0(genes,collapse = "+")))
cox <- coxph(formula,data=lnRNA_expr_survival)
riskScore <- predict(cox,type = "risk",newdata = lnRNA_expr_survival)
expr_survival_riskScore <- lnRNA_expr_survival %>%
mutate(riskScore = as.vector(ifelse(riskScore>median(riskScore),"high","low")))
lnRNA_expr_group <- data.frame(row.names = expr_survival_riskScore$TCGA_full_id,group=expr_survival_riskScore$riskScore)%>%
arrange(desc(group))
lnRNA_expr <- lnRNA_expr[,rownames(lnRNA_expr_group)]
identical(rownames(lnRNA_expr_group),colnames(lnRNA_expr))
deg_expr <- lnRNA_expr[genes,]%>%
{log2(cpm(.)+ 1)}
deg_expr <- t(scale(t(deg_expr)))
deg_expr[deg_expr>2] = 2
deg_expr[deg_expr< -2] = -2
col <- colorRampPalette(c("green","white","red"))(256)
pheatmap(deg_expr,col=col,fontsize=8,show_colnames =F,scale = "row",annotation_col=lnRNA_expr_group,cluster_cols=F)
}
if(F){
## 单基因
my_comparisons <- list( c("i", "ii"))
lnRNA_expr_survival%>%
dplyr::select(c(tumor_stage,LINC00365))%>%
filter(tumor_stage!="unknow") %>%
ggplot(aes(x=tumor_stage,y=LINC00365))+
stat_boxplot(geom="errorbar",width=0.15,aes(color=tumor_stage))+
geom_boxplot(aes(fill=tumor_stage))+
ylim(0, 1000)+
stat_compare_means(comparisons =my_comparisons)+
stat_compare_means(method = "kruskal.test",label = "p.format")
}
mRNA_lnRNA_clinical <- readRDS("result/mRNA_lnRNA_clinical.rda")
mRNA_expr_survival <- mRNA_expr[ce_mRNA,]%>%
t()%>%
as.data.frame()%>%
rownames_to_column("TCGA_full_id")%>%
#filter(substr(TCGA_full_id,14,15)!=11)%>%
mutate(patient_id =substr(TCGA_full_id,1,12))%>%
inner_join(mRNA_lnRNA_clinical,by="patient_id")
Cox_single_res <- Cox_single(mRNA_expr_survival,ce_mRNA,filename = "singleCox_mRNA")
genes <- Cox_single_res%>%filter(pvalue<0.05)%>%pull("id")
res <- Cox_multi(mRNA_expr_survival,genes,filename = "muitiCox_mRNA")
res$outTab
#print(res$image)
Cox_multi_ROC(mRNA_expr_survival,genes,predict.time=5,filename = "muitiCox_mRNA_ROC")
if(F){
if(sum(grepl("-",genes))>0){
genes <- sapply(genes,function(x){sub("-","",x)})
data<- mRNA_expr_survival%>%
rename_all(~sub("-","",.))
}
formula <-as.formula(paste0("Surv(futime_year,fustat)~",paste0(genes,collapse = "+")))
cox <- coxph(formula,data=data)
riskScore <- predict(cox,type = "risk",newdata = data)
expr_survival_riskScore <- data %>%
mutate(riskScore = as.vector(ifelse(riskScore>median(riskScore),"high","low")))
lnRNA_expr_group <- data.frame(row.names = expr_survival_riskScore$TCGA_full_id,group=expr_survival_riskScore$riskScore)%>%
arrange(desc(group))
lnRNA_expr <- lnRNA_expr[,rownames(lnRNA_expr_group)]
identical(rownames(lnRNA_expr_group),colnames(lnRNA_expr))
deg_expr <- lnRNA_expr[genes,]%>%
{log2(cpm(.)+ 1)}
deg_expr <- t(scale(t(deg_expr)))
deg_expr[deg_expr>2] = 2
deg_expr[deg_expr< -2] = -2
col <- colorRampPalette(c("green","white","red"))(256)
pheatmap(deg_expr,col=col,fontsize=8,show_colnames =F,scale = "row",annotation_col=lnRNA_expr_group,cluster_cols=F)
}
if(F){
if(sum(grepl("-",genes))>0){
genes <- sapply(genes,function(x){sub("-","",x)})
lnRNA_expr_survival<- lnRNA_expr_survival%>%
rename_all(~sub("-","",.))
}
formula <-as.formula(paste0("Surv(futime_year,fustat)~",paste0(genes,collapse = "+")))
cox <- coxph(formula,data=lnRNA_expr_survival)
cox <- step(cox,direction = "both")
coxSummary <- summary(cox)
outTab <- data.frame()
outTab <- rbind(outTab,cbind(HR=coxSummary$conf.int[,"exp(coef)"],
HR.95L=coxSummary$conf.int[,"lower .95"],
HR.95H=coxSummary$conf.int[,"upper .95"],
pvalue=coxSummary$coefficients[,"Pr(>|z|)"]))
riskScore <- predict(cox,type = "risk",newdata = lnRNA_expr_survival)
lnRNA_expr_survival_riskScore <- lnRNA_expr_survival %>%
mutate(riskScore = as.vector(ifelse(riskScore< median(riskScore),"high","low")))
fit <- surv_fit(Surv(futime_year,fustat) ~ riskScore, data = lnRNA_expr_survival_riskScore)
ggsurvplot(fit, pval=T, risk.table=F,
risk.table.height = 0.3,data = lnRNA_expr_survival_riskScore,xlab="Time(Years)")
roc <- survivalROC(Stime=lnRNA_expr_survival_riskScore$futime_year ,status=lnRNA_expr_survival_riskScore$fustat,marker=riskScore,
predict.time=5,method="KM")
xyplot(TP~FP,roc,main=paste("ROC curve(","AUC=",round(roc$AUC,3),")"),
xlab="False postive rate",ylab="True positive rate",panel = function(x,y){
panel.xyplot(x,y,type="l",lwd=2,cex.main=1.3,cex.lab=1.2,cex.axis=1.2,font=1.2,col="red")
panel.abline(c(0,1))
})
lnRNA_expr_survival <- lnRNA_expr_survival%>%
select(c(genes,futime_year,fustat))
library(ggrisk)
ggrisk(lnRNA_expr_survival,time="futime_year",event="fustat",heatmap.genes = genes)
}
if(F){
my_comparisons <- list( c("i", "ii"))
mRNA_expr_survival%>%
dplyr::select(c(tumor_stage,CNTN3))%>%
filter(tumor_stage!="unknow") %>%
ggplot(aes(x=tumor_stage,y=CNTN3))+
stat_boxplot(geom="errorbar",width=0.15,aes(color=tumor_stage))+
geom_boxplot(aes(fill=tumor_stage))+
stat_compare_means(comparisons =my_comparisons)+
stat_compare_means(method = "kruskal.test",label = "p.format")
}
#############################################
## ceRNA 子网络构建
#############################################
lnRNA_cox_mutil <- readRDS("result/lnRNA_cox_mutil.rda")
lnRNA_miRNA_mRNA <- readRDS("result/lnRNA_miRNA_mRNA.rda")
lnRNA_miRNA_mRNA_sub <- lnRNA_miRNA_mRNA%>%
filter(lnRNA %in% lnRNA_cox_mutil)
cytoscape(lnRNA_miRNA_mRNA_sub,filename = "small")
saveRDS(lnRNA_miRNA_mRNA_sub,file = "result/sub_netwrok_gene.rda")
if(F){
miRNA_lnRNA_clinical <- readRDS("result/mRNA_lnRNA_clinical.rda")
miRNA_expr <- readRDS("result/miRNA_expr.rda")%>%
rownames_to_column("symbol")%>%
mutate(miRNA_= case_when(grepl("hsa-mir",symbol)~str_extract(symbol,"hsa-mir-[0-9]+[a-z]?"),
grepl("hsa-let",symbol)~str_extract(symbol,"hsa-let-[0-9]+[a-z]?")))%>%
filter(miRNA_ %in% miRNA)%>%
dplyr::select(-miRNA_)%>%
column_to_rownames("symbol")
miRNA_expr_survival <- miRNA_expr%>%
t()%>%
as.data.frame()%>%
rownames_to_column("TCGA_full_id")%>%
mutate(patient_id =substr(TCGA_full_id,1,12))%>%
inner_join(mRNA_lnRNA_clinical,by="patient_id")
Cox_single_res <- Cox_single(miRNA_expr_survival, rownames(miRNA_expr))
genes <- Cox_single_res%>%filter(pvalue<0.05)%>%pull("id")
Cox_multi(mRNA_expr_survival,genes,filename = "ce-lnRNA-Cox_multi")
Cox_multi_ROC(miRNA_expr_survival,predict.time=3,filename = "ce-lnRNA-Cox_multi")
}
##############################################
## GO KEGG
##############################################
(function(){
if(!file.exists("enrichment")){
dir.create(file.path("enrichment"))
}
sub_netwrok_gene <- readRDS("result/sub_netwrok_gene.rda")
if(F){
gene <- unique(sub_netwrok_gene[sub_netwrok_gene$miRNA=="hsa-mir-34b",]$mRNA)
write.csv(gene,file = "result/GO_KEKK_GENE.csv",quote = F,row.names = F)
}
gene <- unique(sub_netwrok_gene$mRNA)
entrez_id <- bitr(gene, fromType = "SYMBOL",
toType = c("ENTREZID"),
OrgDb = org.Hs.eg.db)%>%
pull("ENTREZID")%>%unique()
egobp_BP<-enrichGO(gene = entrez_id,OrgDb = org.Hs.eg.db,keyType = 'ENTREZID',ont = 'BP',pAdjustMethod="none") # BP MF CC ALL
#dotplot(egobp_BP,showCategory=20)
egobp_BP@result%>%
arrange(p.adjust)%>%
write.csv(file = "result/enrichment/egobp_BP.csv",quote = F,row.names = F)
write.csv(egobp_BP@result)
egobp_MF<-enrichGO(gene = entrez_id,OrgDb = org.Hs.eg.db,keyType = 'ENTREZID',ont = 'MF',pAdjustMethod="none") # BP MF CC ALL
#dotplot(egobp_MF,showCategory=20)
egobp_MF@result%>%
arrange(p.adjust)%>%
write.csv(file = "result/enrichment/egobp_MF.csv",quote = F,row.names = F)
egobp_CC<-enrichGO(gene = entrez_id,OrgDb = org.Hs.eg.db,keyType = 'ENTREZID',ont = 'CC',pAdjustMethod="none") # BP MF CC ALL
#dotplot(egobp_CC,showCategory=20)
egobp_CC@result%>%
arrange(p.adjust)%>%
write.csv(file = "result/enrichment/egobp_CC.csv",quote = F,row.names = F)
kk <- clusterProfiler::enrichKEGG(gene =entrez_id,organism = 'hsa',pvalueCutoff = 0.4,qvalueCutoff = 0.3,pAdjustMethod="none")
#dotplot(kk,showCategory=20)
kk@result%>%
arrange(p.adjust)%>%
write.csv(file = "result/enrichment/kegg.csv",quote = F,row.names = F)
})()