使用R包下载TCGA数据
TCGAbiolinks
browseVignettes("TCGAbiolinks") 查看帮助
下载count数据
library(TCGAbiolinks)
GDC_projects <- getGDCprojects() # 查看TCGA项目
GDC_projects
# 查询数据
query_LIHC_counts <- GDCquery(project = "TCGA-LIHC",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts")
# 下载数据
GDCdownload(query_LIHC_counts)
# 合并数据
LIHC_rnaseq_biolinks <- GDCprepare(query = query_LIHC_counts,
summarizedExperiment = FALSE,
save = TRUE,
save.filename = "data/LIHC_TCGAbiolinks/LIHC_rnaseq_biolinks.rda")
load(file = "data/LIHC_TCGAbiolinks/LIHC_rnaseq_biolinks.rda")
tail(LIHC_rnaseq_biolinks, 10)[ ,1:5]
summarizedExperiment = T
library(SummarizedExperiment)
summary(RT_rnaseq_SE)
col_data <- colData(RT_rnaseq_SE)
row_data <- rowData(RT_rnaseq_SE)
expr_data <- assay(RT_rnaseq_SE)

图片alt
下载临床信息
# clinical data
query_clin_LIHC <- GDCquery(project = "TCGA-LIHC",
data.category = "Clinical",
data.type = "Clinical Supplement",
data.format = "BCR Biotab")
# GDCdownload(query_clin_LIHC)
clinical.BCRtab.all <- GDCprepare(query_clin_LIHC)
names(clinical.BCRtab.all)
sort(colnames(clinical.BCRtab.all$clinical_patient_lihc))

图片alt
# 方式二
LIHC_clin <- GDCquery_clinic(project = "TCGA-LIHC", type = "clinical")
LIHC_clin_xena <- readr::read_tsv("data/LIHC_UCSC/TCGA-LIHC.GDC_phenotype.tsv.gz")
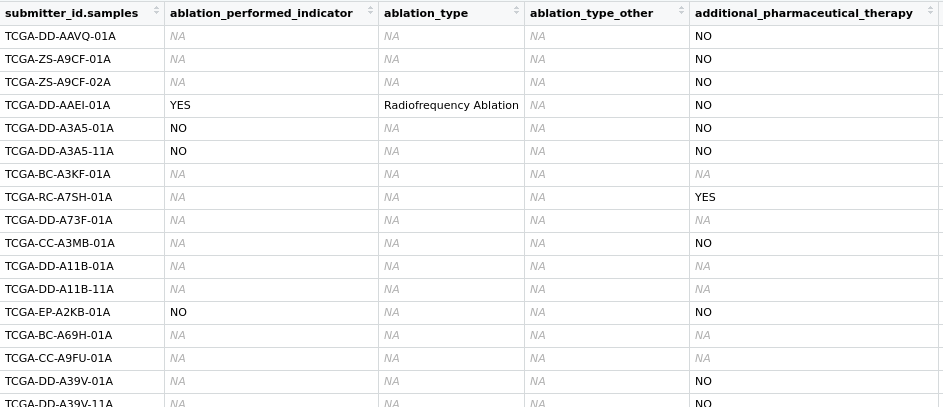
图片alt

图片alt
TCGA数据预处理
TCGA构建metadata信息

图片alt

图片alt
json_file <- "data/LIHC_GDC/metadata.cart.2020-07-15.json"
TCGA_metadata <- function(path = json_file) {
metadata <- jsonlite::read_json(path, simplifyVector = T)
metadata <- tibble::tibble(
file_name = metadata$file_name,
md5sum = metadata$md5sum,
TCGA_id_full = bind_rows(metadata$associated_entities)$entity_submitter_id,
TCGA_id = stringr::str_sub(TCGA_id_full, 1, 16),
patient_id = stringr::str_sub(TCGA_id, 1, 12),
tissue_type_id = stringr::str_sub(TCGA_id, 14, 15),
tissue_type = sapply(tissue_type_id, function(x) {
switch(x,
"01" = "Primary Solid Tumor",
"02" = "Recurrent Solid Tumor",
"03" = "Primary Blood Derived Cancer - Peripheral Blood",
"05" = "Additional - New Primary",
"06" = "Metastatic",
"07" = "Additional Metastatic",
"11" = "Solid Tissue Normal")}),
group = ifelse(tissue_type_id == "11", "Normal", "Tumor"))
return(metadata)
}
metadata2 <- TCGA_metadata()

图片alt
从本地下载count数据
file_path <- "data/LIHC_GDC/gdc_download_20200715_050611.695593"
counts_files <- list.files(path = file_path,
pattern = "counts.gz",
full.names = T,
recursive = T)
head(counts_files)
# 验证下载文件的md5sum值
pkgs_in("tools")
all(tools::md5sum(counts_files) %in% metadata$md5sum)
## first file
test <- readr::read_tsv(counts_files[1], col_names = F)
head(test)
tail(test)
## merged counts data
counts_df <- counts_files %>%
lapply(function(x) {
tmp <- read_tsv(x, col_names = F) %>%
purrr::set_names("gene_id", basename(x))
cat(which(counts_files == x), "of", length(counts_files), "\n")
return(tmp)
}) %>%
reduce(function(x, y) full_join(x, y, by = "gene_id")) %>%
dplyr::select(gene_id, metadata$file_name) %>%
set_names("gene_id", metadata$TCGA_id_full) %>%
dplyr::slice(1:(nrow(.)-5))
LIHC_counts <- counts_df
LIHC_metadata <- metadata
save(LIHC_counts, LIHC_metadata, file = "data/LIHC_GDC/LIHC_counts.rda")
TCGA测序数据ID装换
gtf_v22 <- read_tsv("data/gencode.gene.info.v22.tsv")
id2symbol <- gtf_v22 %>%
dplyr::select(1, 2)
LIHC_counts_filtered_anno <- id2symbol %>%
inner_join(LIHC_counts_filtered, by = "gene_id")

图片alt
TCGA数据过滤
样本过滤
sample_anno <- readr::read_tsv(file = "data/merged_sample_quality_annotations.tsv")
LIHC_anno <- sample_anno %>%
dplyr::filter(aliquot_barcode %in% colnames(LIHC_counts)) %>%
write_tsv("data/LIHC_anno.tsv")
sample_good <- read_tsv("data/LIHC_anno_examined.tsv") %>%
pull(aliquot_barcode) %>%
na.omit()
sample_rm <- setdiff(LIHC_anno$aliquot_barcode, sample_good)
LIHC_counts_filtered <- LIHC_counts %>%
select(gene_id, all_of(sample_good))

图片alt
基因过滤
- 去除表达量全部为0的基因
- 保留至少在60%样本中表达量大于0的基因
- 保留中位数大于0的基因
- 保留至少在一个最小分组的样本数量中表达的基因
提取mRNA及lncRNA表达矩阵
gtf_v22 <- read_tsv("data/gencode.gene.info.v22.tsv")
## lncRNA
sort(table(gtf_v22$gene_type), decreasing = T)
sort(unique(gtf_v22$gene_type))
## lncRNA 范围 from ensembl.org
## https://m.ensembl.org/info/genome/genebuild/biotypes.html
# Long non-coding RNA (lncRNA): A non-coding gene/transcript >200bp in length
# 3' overlapping ncRNA: Transcripts where ditag and/or published experimental data strongly supports the existence of long (>200bp) non-coding transcripts that overlap the 3'UTR of a protein-coding locus on the same strand.
# Antisense: Transcripts that overlap the genomic span (i.e. exon or introns) of a protein-coding locus on the opposite strand.
# Macro lncRNA: Unspliced lncRNAs that are several kb in size.
# Non coding: Transcripts which are known from the literature to not be protein coding.
# Retained intron: An alternatively spliced transcript believed to contain intronic sequence relative to other, coding, transcripts of the same gene.
# Sense intronic: A long non-coding transcript in introns of a coding gene that does not overlap any exons.
# Sense overlapping: A long non-coding transcript that contains a coding gene in its intron on the same strand.
# lincRNA (long intergenic ncRNA): Transcripts that are long intergenic non-coding RNA locus with a length >200bp. Requires lack of coding potential and may not be conserved between species.
lncRNA_types <- "3prime_overlapping_ncrna, antisense, bidirectional_promoter_lncRNA, lincRNA, macro_lncRNA, non_coding, processed_transcript, sense_intronic, sense_overlapping"
lncRNA_types <- unlist(str_split(lncRNA_types, pattern = ", "))
lncRNA_types
lncRNA_types %in% unique(gtf_v22$gene_type)
## gene type info
gene_type <- gtf_v22 %>%
select(gene_id, gene_type) %>%
arrange(gene_id)
dim(gene_type)
identical(gene_type$gene_id, LIHC_counts$gene_id)
## 提取mRNA
expr_mRNA <- LIHC_counts[gene_type$gene_type == "protein_coding", ]
## 提取lncRNA
expr_lncRNA <- LIHC_counts[gene_type$gene_type %in% lncRNA_types, ]
TCGA和GTEx数据合并
作用弥补TCGA数据库中缺少癌旁组织样
TCGA miRNA测序数据整理
#### 1 miRNA expression ----
## import json file
source("codes/custom_functions.R")
pkgs_in("jsonlite")
library(tidyverse)
json_file <- "data/LIHC_miRNA/miRNA_Expression/metadata.cart.LIHC.miRNA.json"
metadata <- TCGA_metadata()
## miRNA_files
file_path <- "data/LIHC_miRNA/miRNA_Expression/"
miRNA_files <- list.files(path = file_path,
pattern = "mirnas.quantification.txt",
full.names = T,
recursive = T)
head(miRNA_files)
## 验证下载文件的md5sum值
pkgs_in("tools")
all(tools::md5sum(miRNA_files) %in% metadata$md5sum)
## first file
test <- readr::read_tsv(miRNA_files[1])
dim(test)
head(test)
tail(test)
tail(test$read_count/sum(test$read_count)*10^6)
## merged miRNA counts data
miRNA_df <- miRNA_files %>%
lapply(function(x) {
tmp <- read_tsv(x, col_names = T)[ ,1:2] %>%
purrr::set_names("miRNA_id", basename(x))
cat(which(miRNA_files == x), "of", length(miRNA_files), "\n")
return(tmp)
}) %>%
purrr::reduce(.f = function(x, y) full_join(x, y, by = "miRNA_id")) %>%
dplyr::select(miRNA_id, metadata$file_name) %>%
purrr::set_names("miRNA_id", metadata$TCGA_id_full)
sum(is.na(miRNA_df))
## save data
LIHC_miRNA <- miRNA_df
LIHC_metadata_miRNA <- metadata
save(LIHC_miRNA, LIHC_metadata_miRNA, file = "data/LIHC_miRNA/LIHC_miRNA.rda")
#### 2 miRNA isoform expression ----
## import json file
rm(list = ls())
source("codes/custom_functions.R")
pkgs_in("jsonlite")
library(tidyverse)
json_file <- "data/LIHC_miRNA/miRNA_Isoform/metadata.cart.LIHC.miRNA.Isoform.json"
metadata <- TCGA_metadata()
## miRNA_isoform_files
file_path <- "data/LIHC_miRNA/miRNA_Isoform/"
miRNA_isoform_files <- list.files(path = file_path,
pattern = "isoforms.quantification.txt",
full.names = T,
recursive = T)
head(miRNA_isoform_files, 3)
## 验证下载文件的md5sum值
pkgs_in("tools")
all(tools::md5sum(miRNA_isoform_files) %in% metadata$md5sum)
## first file
test <- readr::read_tsv(miRNA_isoform_files[1], col_names = T)
head(test)
tail(test)
## miRNA注释信息
# ftp://mirbase.org/pub/mirbase/21/
miRNA_anno <- read_lines("data/LIHC_miRNA/mature.fa.gz") %>%
as_tibble() %>%
dplyr::filter(str_detect(value, pattern = "Homo")) %>%
tidyr::separate(col = value, into = paste0("ID", 1:5), sep = " ") %>%
dplyr::select(isoform_id = ID2, miRNA_name = ID5)
## merged miRNA isoform counts data
isoform_list <- miRNA_isoform_files %>%
lapply(function(x) {
tmp <- read_tsv(x) %>%
dplyr::select(isoform_id = miRNA_region, read_count) %>%
mutate(isoform_id = str_extract(isoform_id, "MIMAT.*")) %>%
na.omit() %>%
aggregate(. ~ isoform_id, data = ., sum) %>%
# aggregate(read_count ~ isoform_id, data = ., sum) %>%
purrr::set_names("isoform_id", basename(x))
cat(which(miRNA_isoform_files == x), "of", length(miRNA_isoform_files), "\n")
return(tmp)
})
## aggregate
# aggregate(weight ~ feed, data = chickwts, mean)
# aggregate(breaks ~ wool + tension, data = warpbreaks, mean)
# aggregate(cbind(Ozone, Temp) ~ Month, data = airquality, mean)
# aggregate(cbind(ncases, ncontrols) ~ alcgp + tobgp, data = esoph, sum)
# iris
# aggregate(. ~ Species, data = iris, mean)
# ToothGrowth
# aggregate(len ~ ., data = ToothGrowth, mean)
test2 <- full_join(x = isoform_list[[1]],
y = isoform_list[[2]],
by = "isoform_id")
isoform_df <- isoform_list %>%
reduce(function(x, y) full_join(x, y, by = "isoform_id")) %>%
select(isoform_id, metadata$file_name) %>%
set_names("isoform_id", metadata$TCGA_id_full) %>%
mutate(across(.fns = ~ ifelse(is.na(.x), 0, .x))) %>%
right_join(miRNA_anno, ., by = "isoform_id")
sum(is.na(isoform_df))
## save data
LIHC_miRNA_iso <- isoform_df
LIHC_metadata_miRNA_iso <- metadata
save(LIHC_miRNA_iso, LIHC_metadata_miRNA_iso,
file = "data/LIHC_miRNA/LIHC_miRNA_isoform.rda")
#### End ----
mircode数据整理
get_mircode <- function(){
if(!file.exists("result/mircode.rda")){
mircode <- read.table("/home/wangyang/workspace/bioinfo_analysis/Rscript/data/mircode_highconsfamilies.txt",sep = "\t",header = T)
table(mircode$gene_class)
lncmiRcode = mircode[mircode$gene_class %in%c("lncRNA, intergenic","lncRNA, overlapping"),1:4]
head(lncmiRcode$microrna)
library(stringr)
p1 = str_starts(lncmiRcode$microrna,"miR-")
table(p1)
p2 = str_starts(lncmiRcode$microrna,"let-")
table(p2)
lncmiRcode_let <- lncmiRcode[p2,]%>%
dplyr::select(c("gene_symbol","microrna"))%>%
mutate(miRNA=str_replace_all(microrna,"let-",""))%>%
separate_rows(miRNA,sep = "/",convert = T)%>%
mutate_at(vars(contains("miRNA")), ~ paste0("hsa-let-",.))%>%
mutate(miRNA = str_extract(miRNA,"hsa-let-[0-9]+[a-z]?"))%>%
dplyr::select(c("gene_symbol","miRNA"))
lncmiRcode_miR <- lncmiRcode[p1,]%>%
dplyr::select(c("gene_symbol","microrna"))%>%
mutate(miRNA=str_replace_all(microrna,"miR-",""))%>%
separate_rows(miRNA,sep = "/",convert = T)%>%
mutate_at(vars(contains("miRNA")), ~ paste0("hsa-mir-",.))%>%
mutate(miRNA = str_extract(miRNA,"hsa-mir-[0-9]+[a-z]?"))%>%
dplyr::select(c("gene_symbol","miRNA"))
lncmiRcode_f <- rbind(lncmiRcode_let,lncmiRcode_miR)
saveRDS(lncmiRcode_f,file = "result/mircode.rda")
return(lncmiRcode_f)
}
return(readRDS("result/mircode.rda"))
}
TCGA数据质量评估
#### load data ----
load_data <- load("data/LIHC_GDC/LIHC_counts.rda")
#### 样本过滤 ----
# Merged Sample Quality Annotations - merged_sample_quality_annotations.tsv
# https://gdc.cancer.gov/about-data/publications/pancanatlas
library(tidyverse)
sample_anno <- readr::read_tsv(file = "data/merged_sample_quality_annotations.tsv")
LIHC_anno <- sample_anno %>%
dplyr::filter(aliquot_barcode %in% colnames(LIHC_counts)) %>%
write_tsv("data/LIHC_anno.tsv")
sample_good <- read_tsv("data/LIHC_anno_examined.tsv") %>%
pull(aliquot_barcode) %>%
na.omit()
sample_rm <- setdiff(LIHC_anno$aliquot_barcode, sample_good)
LIHC_counts_filtered <- LIHC_counts %>%
select(gene_id, all_of(sample_good))
#### 准备数据 ----
### metadata ----
metadata <- LIHC_metadata %>%
filter(tissue_type_id %in% c("01", "11")) %>%
add_count(patient_id, name = "n_patient") %>%
filter(n_patient == 2) %>%
arrange(patient_id) %>%
as.data.frame()
rownames(metadata) <- metadata$TCGA_id
### counts ----
paired_counts <- LIHC_counts %>%
set_names("gene_id", str_sub(colnames(.)[-1], 1,16)) %>%
column_to_rownames("gene_id") %>%
select(metadata$TCGA_id)
keep <- rowSums(paired_counts > 0) >= 50
head(keep)
paired_counts <- paired_counts[keep,]
### vst ----
library(DESeq2)
group <- metadata$group %>% as.factor()
colData = data.frame(sample_id = colnames(paired_counts), group = group)
DDS <- DESeq2::DESeqDataSetFromMatrix(paired_counts,
colData = metadata,
design = ~ group)
vst <- DESeq2::vst(DDS)
counts_vst <- assay(vst)
#### boxplot & density plot ----
boxplot(log2(paired_counts+1), las = 2, outline = F, col = c("red", "blue"))
limma::plotDensities(log2(paired_counts+1), legend = F)
boxplot(counts_vst, las = 2, outline = F, col = group)
limma::plotDensities(counts_vst , legend = F)
## ggplot2 data
data_counts <- log2(paired_counts + 1) %>%
pivot_longer(cols = everything(), names_to = "TCGA_id") %>%
left_join(metadata %>% select(TCGA_id, group), by = "TCGA_id")
data_vst <- counts_vst %>%
as.data.frame() %>%
pivot_longer(cols = everything(), names_to = "TCGA_id") %>%
left_join(metadata %>% select(TCGA_id, group), by = "TCGA_id")
## ggplot2 box plot
ggplot(data_counts, aes(x = TCGA_id, y = value, fill = group)) +
geom_boxplot(outlier.shape = NA) +
theme_bw() +
xlab("samples") + ylab("log2 count value") +
theme(axis.text.x = element_blank(),
axis.ticks = element_blank())
ggplot(data_vst, aes(x = TCGA_id, y = value, fill = group)) +
geom_boxplot(outlier.shape = NA) +
theme_bw() +
xlab("samples") + ylab("vst transformed value") +
theme(axis.text.x = element_blank(),
axis.ticks = element_blank())
## ggplot2 density plot
ggplot(data_counts, aes(x = value, col = TCGA_id)) +
geom_density() +
theme_bw() +
xlab("samples") +
xlim(0, 20) +
theme(legend.position = "none")
ggplot(data_vst, aes(x = value, col = TCGA_id)) +
geom_density() +
theme_bw() +
xlim(0, 20) +
theme(legend.position = "none")
sample_rm <- str_sub(sample_rm, 1, 16)
sum(colnames(counts_vst) %in% sample_rm)
data_vst_filter <- data_vst %>%
dplyr::filter(!(TCGA_id %in% sample_rm))
ggplot(data_vst_filter, aes(x = value, col = TCGA_id)) +
geom_density() +
theme_bw() +
xlim(0, 20) +
theme(legend.position = "none")
#### PCA plot ----
source("codes/custom_functions.R")
PCA_new(log2(paired_counts+1), group = metadata$group)
PCA_new(counts_vst, group = metadata$group)
# 加载包
library("factoextra")
library("FactoMineR")
# 准备数据
pca <- PCA(t(log2(paired_counts+1)), graph = FALSE)
# 作图
fviz_pca_ind(pca,
label = "none",
habillage = as.factor(metadata$group),
palette = c("#00AFBB", "#E7B800"),
addEllipses = TRUE)
#### 聚类树状图 ----
expr <- as.matrix(counts_vst)
dist <- dist(t(expr))
hc = hclust(dist)
library(factoextra)
library(RColorBrewer)
pdf("data/clust.pdf", height = 10, width = 20)
fviz_dend(hc, k = 6,
cex = 0.5,
k_colors = brewer.pal(4, "Set2"),
color_labels_by_k = TRUE,
ggtheme = theme_classic())
dev.off()
#### End ----
差异基因分析
从TCGAbiolinks获得分组信息
metadata <- query_COAD_counts[[1]][[1]]
metadata <- tibble::tibble(
TCGA_id_full = metadata$cases,
TCGA_id = stringr::str_sub(TCGA_id_full, 1, 16),
patient_id = stringr::str_sub(TCGA_id, 1, 12),
tissue_type_id = stringr::str_sub(TCGA_id, 14, 15),
tissue_type = sapply(tissue_type_id, function(x) {
switch(x,
"01" = "Primary Solid Tumor",
"02" = "Recurrent Solid Tumor",
"03" = "Primary Blood Derived Cancer - Peripheral Blood",
"05" = "Additional - New Primary",
"06" = "Metastatic",
"07" = "Additional Metastatic",
"11" = "Solid Tissue Normal")}),
group = ifelse(tissue_type_id == "11", "Normal", "Tumor"))
rownames(metadata) <- metadata$TCGA_id_full
从json文件获得分组信息
TCGA_metadata <- function(path = json_file) {
metadata <- jsonlite::read_json(path, simplifyVector = T)
metadata <- tibble::tibble(
file_name = metadata$file_name,
md5sum = metadata$md5sum,
TCGA_id_full = bind_rows(metadata$associated_entities)$entity_submitter_id,
TCGA_id = stringr::str_sub(TCGA_id_full, 1, 16),
patient_id = stringr::str_sub(TCGA_id, 1, 12),
tissue_type_id = stringr::str_sub(TCGA_id, 14, 15),
tissue_type = sapply(tissue_type_id, function(x) {
switch(x,
"01" = "Primary Solid Tumor",
"02" = "Recurrent Solid Tumor",
"03" = "Primary Blood Derived Cancer - Peripheral Blood",
"05" = "Additional - New Primary",
"06" = "Metastatic",
"07" = "Additional Metastatic",
"11" = "Solid Tissue Normal")}),
group = ifelse(tissue_type_id == "11", "Normal", "Tumor"))
return(metadata)
}
metadata2 <- TCGA_metadata()
count数据整理
counts <- LIHC_counts_filtered %>%
column_to_rownames("gene_id") %>%
dplyr::select(metadata$TCGA_id_full)
identical(colnames(counts), rownames(metadata))
DESeq2 差异表达分析
### 2.1 构建dds对象 ----
library(DESeq2)
?DESeqDataSetFromMatrix()
dds <- DESeqDataSetFromMatrix(countData = counts,
colData = metadata,
design = ~ group)
## PCA图
vsd <- vst(dds, blind = TRUE)
vsd_df <- assay(vsd)
head(vsd_df, 5)[, 1:2]
DESeq2::plotPCA(vsd, intgroup = "group")
source("codes/custom_functions.R")
PCA_new(expr = vsd_df, group = metadata$group)
## remove batch effect
vsd_batch_rm <- limma::removeBatchEffect(x = vsd_df, batch = metadata$batch)
PCA_new(expr = vsd_batch_rm, group = metadata$group)
### 2.2 基因过滤 ----
table(metadata$group)
keep <- rowSums(counts(dds) > 0) >= 44
dds_filt <- dds[keep, ]
### 2.3 DESeq一步完成差异分析 ----
time1 <- Sys.time()
dds2 <- DESeq(dds_filt)
runtime1 <- Sys.time() - time1
runtime1 # Time difference of 4.238453 mins
time2 <- Sys.time()
dds2 <- DESeq(dds_filt, parallel = T)
runtime2 <- Sys.time() - time2
runtime2 # Time difference of 3.506404 mins
# save(dds2, file = "data/dds2.Rda")
load(file = "data/dds2.Rda")
### 2.4 提取差异分析结果 ----
resultsNames(dds2)
res <- results(dds2)
# 基因ID对应的基因名
gtf_v22 <- read_tsv("data/gencode.gene.info.v22.tsv") %>%
dplyr::select(gene_id, gene_name)
res_deseq2 <- as.data.frame(res) %>%
rownames_to_column("gene_id") %>%
left_join(gtf_v22, by = "gene_id") %>%
relocate(gene_name, .after = "gene_id") %>%
arrange(padj) %>%
dplyr::filter(abs(log2FoldChange) > 1, padj < 0.05)
# Note on p-values set to NA: some values in the results table can be set to NA for one of the following reasons:
# If within a row, all samples have zero counts, the baseMean column will be zero, and the log2 fold change estimates, p value and adjusted p value will all be set to NA.
# If a row contains a sample with an extreme count outlier then the p value and adjusted p value will be set to NA. These outlier counts are detected by Cook’s distance.
# If a row is filtered by automatic independent filtering, for having a low mean normalized count, then only the adjusted p value will be set to NA.
# baseMean
# https://support.bioconductor.org/p/75244/
head(rowMeans(DESeq2::counts(dds2, normalized=TRUE, replaced = TRUE)))
纳入 batch effect 进行差异表达分析
dds_batch <- DESeqDataSetFromMatrix(countData = counts,
colData = metadata,
design = ~ batch + group)
keep <- rowSums(counts(dds_batch) > 0) >= 44
dds_filt2 <- dds_batch[keep, ]
dds_batch2 <- DESeq2::DESeq(dds_filt2, parallel = T)
# save(dds_batch2, file = "data/dds_batch2.Rda")
load("data/dds_batch2.Rda")
resultsNames(dds_batch2)
res_batch <- results(dds_batch2)
res_deseq2_batch <- as.data.frame(res_batch) %>%
rownames_to_column("gene_id") %>%
left_join(gtf_v22, by = "gene_id") %>%
relocate(gene_name, .after = "gene_id") %>%
arrange(padj) %>%
dplyr::filter(abs(log2FoldChange) > 1, padj < 0.05)
batch effect 去除前后差异分析结果比较
length(intersect(res_deseq2_batch$gene_id, res_deseq2$gene_id))
library(VennDiagram)
venn.diagram(x = list("rm_batch" = res_deseq2_batch$gene_id,
"batch" = res_deseq2$gene_id),
filename = "results/Venn_2.jpeg",
fill = c("blue", "red"),
scaled = T,
cex = 1.5,
cat.cex = 1.5,
main = "Venn Diagram",
main.cex = 2)
TCGA数据批量下载
library(TCGAbiolinks)
project_df <- (function(){
projects <- TCGAbiolinks:::getGDCprojects()$project_id
projects_full <- projects[grepl('^TCGA',projects,perl=T)]
projects_ID <- sapply(projects_full,function(x){stringi::stri_sub(x,6,10)})
res <- data.frame(cancer=projects_ID,TCGA_ID=projects_full)
return(res)
})()
readr::write_csv(project_df,file = "TCGA_PROJECT.csv")
import pandas as pd
df = pd.read_csv("TCGA_PROJECT.csv")
for item in df['TCGA_ID']:
print("./DownloadFPKM.R FPKM {0}".format(item))
print("./DownloadFPKM.R Counts {0}".format(item))
print("./DownloadFPKM.R miRNA {0}".format(item))
#!/usr/bin/Rscript
library(TCGAbiolinks)
getwd()
args=commandArgs(T)
print(args[1])
#type="FPKM"
#project = "TCGA-CHOL"
type=args[1]
project = args[2]
#####下载FPKM
(function(){
if(!file.exists("GDCdata/FPKM")){
dir.create("GDCdata/FPKM",recursive = T)
}
if(!file.exists("GDCdata/Counts")){
dir.create("GDCdata/Counts",recursive = T)
}
if(!file.exists("result")){
dir.create("result")
}
if(!file.exists("GDCdata/miRNA")){
dir.create("GDCdata/miRNA",recursive = T)
}
# match.file.cases.all <- NULL
# for(project in project_df[,2]){
filename <- gsub("-","_",paste0("result/",project,"_FPKM.tsv"))
if(file.exists(filename)){
message("###########",filename,"存在#######################################################")
}else{
if(type=="FPKM"){
query<- GDCquery(project = project,
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - FPKM")
GDCdownload(query,directory = "GDCdata/FPKM",method = "api")
data <- GDCprepare(query,
directory = "GDCdata/FPKM",
summarizedExperiment = F,
save = F)
readr::write_tsv(data,file =filename )
}else if(type=="Counts"){
query<- GDCquery(project = project,
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts")
GDCdownload(query,directory = "GDCdata/Counts",method = "api")
data <- GDCprepare(query,
directory = "GDCdata/Counts",
summarizedExperiment = F,
save = F)
readr::write_tsv(data,file =filename )
}else if(type=="miRNA"){
query<- GDCquery(project = project,
data.category = "Transcriptome Profiling",
data.type = "miRNA Expression Quantification",
workflow.type = "BCGSC miRNA Profiling")
GDCdownload(query,directory = "GDCdata/miRNA",method = "api")
data <- GDCprepare(query,
directory = "GDCdata/miRNA",
summarizedExperiment = F,
save = F)
readr::write_tsv(data,file =filename )
}
}
})()
#!/usr/bin/Rscript
library(TCGAbiolinks)
getwd()
project_df <- (function(){
projects <- TCGAbiolinks:::getGDCprojects()$project_id
projects_full <- projects[grepl('^TCGA',projects,perl=T)]
projects_ID <- sapply(projects_full,function(x){stringi::stri_sub(x,6,10)})
res <- data.frame(cancer=projects_ID,TCGA_ID=projects_full)
return(res)
})()
#####下载FPKM
(function(){
if(!file.exists("GDCdata/FPKM")){
dir.create("GDCdata/FPKM",recursive = T)
}
if(!file.exists("result")){
dir.create("result")
}
match.file.cases.all <- NULL
for(project in project_df[,2]){
filename <- gsub("-","_",paste0("result/",project,"_FPKM.tsv"))
if(file.exists(filename)){
message("###########",filename,"存在#######################################################")
next
}else{
query<- GDCquery(project = project,
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - FPKM")
match.file.cases <- getResults(query,cols=c("cases","file_name"))
match.file.cases$project <- project
match.file.cases.all <- rbind(match.file.cases.all,match.file.cases)
GDCdownload(query,directory = "GDCdata/FPKM",method = "api")
table(match.file.cases.all$project)
data <- GDCprepare(query,
directory = "GDCdata/FPKM",
summarizedExperiment = F,
save = F)
readr::write_tsv(data,file =filename )
}
}
readr::write_tsv(match.file.cases.all, path = "TCGA_FPKM.tsv")
})()
#####下载COUNT
(function(){
if(!file.exists("GDCdata/Counts")){
dir.create("GDCdata/Counts",recursive = T)
}
if(!file.exists("result")){
dir.create("result")
}
match.file.cases.all <- NULL
for(project in project_df[,2]){
filename <- gsub("-","_",paste0("result/",project,"_Counts.tsv"))
if(file.exists(filename)){
message("###########",filename,"存在#######################################################")
next
}else{
query<- GDCquery(project = project,
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts")
match.file.cases <- getResults(query,cols=c("cases","file_name"))
match.file.cases$project <- project
match.file.cases.all <- rbind(match.file.cases.all,match.file.cases)
GDCdownload(query,directory = "GDCdata/Counts",method = "api")
table(match.file.cases.all$project)
data <- GDCprepare(query,
directory = "GDCdata/Counts",
summarizedExperiment = F,
save = F)
readr::write_tsv(data,file =filename )
}
# project <- "TCGA-BRCA"
}
readr::write_tsv(match.file.cases.all, path = "TCGA_Counts.tsv")
})()
#####下载miRNA
(function(){
if(!file.exists("GDCdata/miRNA")){
dir.create("GDCdata/miRNA",recursive = T)
}
if(!file.exists("result")){
dir.create("result")
}
match.file.cases.all <- NULL
for(project in project_df[,2]){
filename <- gsub("-","_",paste0("result/",project,"_miRNA.tsv"))
if(file.exists(filename)){
message("###########",filename,"存在#######################################################")
next
}else{
#project <- "TCGA-BRCA"
query<- GDCquery(project = project,
data.category = "Transcriptome Profiling",
data.type = "miRNA Expression Quantification",
workflow.type = "BCGSC miRNA Profiling")
match.file.cases <- getResults(query,cols=c("cases","file_name"))
match.file.cases$project <- project
match.file.cases.all <- rbind(match.file.cases.all,match.file.cases)
GDCdownload(query,directory = "GDCdata/miRNA",method = "api")
table(match.file.cases.all$project)
data <- GDCprepare(query,
directory = "GDCdata/miRNA",
summarizedExperiment = F,
save = F)
readr::write_tsv(data,file =filename )
}
}
readr::write_tsv(match.file.cases.all, path = "TCGA_miRNA.tsv")
})()